Dalle prime idee sulla costituzione atomica e sulle forze interatomiche alla struttura dell’atomo e delle molecole secondo la fisica dei quanti.
PARTE 2: MOLECOLE
Piano della Parte 2
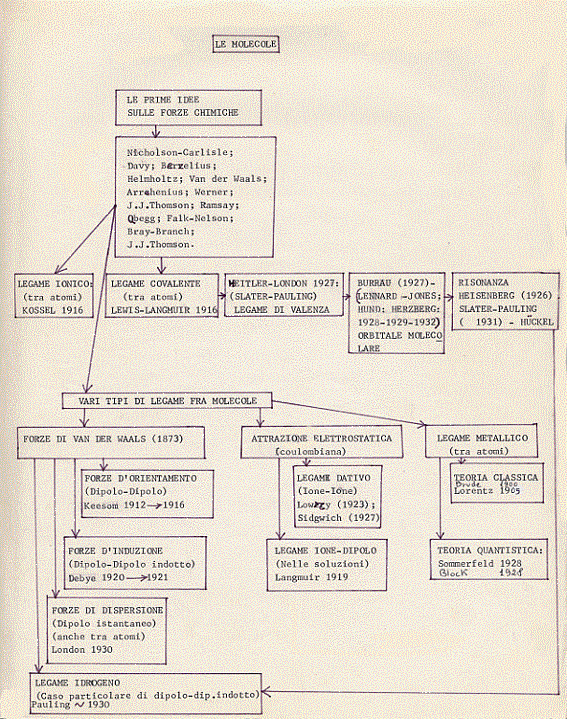
1 – Introduzione
Abbiamo studiato gli atomi. Ma la gran parte delle sostanze che ci circondano sono formate da gruppi di atomi (o molecole), scindendo i quali si hanno delle sostanze con delle caratteristiche diverse da quelle della sostanza considerata.
Come abbiamo già ricordato fu Democrito il primo ad intuire che la materia fosse composta da atomi. Ma Democrito credeva che ogni sostanza diversa esistente fosse formata da atomi di diverso tipo: tante sostanze diverse, tanti atomi diversi. Un contemporaneo di Democrito, Empedocle, sviluppò ulteriormente l’idea dello stesso Democrito. Egli sosteneva che vi sono solo poche specie di atomi diversi, i quali, combinandosi insieme in vari modi originavano tutti gli oggetti che ci è possibile osservare nell’universo. Empedocle riteneva che solo quattro fossero le specie di atomi costituenti tutte le sostanze: atomi di terra, atomi di aria, atomi di fuoco ed atomi di acqua.
Questi atomi erano tenuti insieme dall’amore, si disgregavano a causa dell’odio. Queste due entità, amore ed odio, governavano tutti i cam pi delle umane attività.
Queste idee vennero in qualche modo riprese dagli alchimisti alessandrini (~ 300 d.C.) e dagli alchimisti dell’Islam orientale (~ 800 d.C.). Le premesse fondamentali dell’alchimia, che hanno probabilmente origine alessandrina (~ 100 d.C.) si possono così riassumere:
1) Tutte le materie sono formate da alcuni componenti, i quattro elementi di Empedocle, in varie mescolanze tra loro.
2) Vi è una scala di nobiltà e purezza degli elementi. Il più nobile è l’oro; segue l’argento.
3) E’ possibile che un metallo si trasformi in un altro metallo mutando il rapporto quantitativo dei quattro elementi che lo costituiscono.
4) Si può ottenere un metallo nobile da uno vile, impregnando il metallo vile di una sostanza preziosa spesso chiamata il quinto elemento (quintessenza) o elisir.
Il lavoro degli alchimisti consistette in una serie di osservazioni pratiche sulla formazione di vari composti ottenuti miscelando, riscal dando, distillando e con altri procedimenti.
Gradualmente furono isolate e catalogate circa 90 sostanze (elementi) che non potevano essere decomposte ulteriormente e furono descritte centinaia di sostanze (composti) ottenute come combinazione delle prime.
Nasce così la prima definizione di molecola (da molecula = piccola massa. Il nome ed il concetto di molecola furono coniati intorno al 1618 dal medico francese Daniel Sennert): la più piccola par te di una sostanza che ha in sé tutte le proprietà della sostanza.
Una ulteriore estensione di questa idea a molecole formate da atomi della stessa specie (H2 , N2 , O2 …) fu fatta da Avogadro come del resto abbiamo già detto nella prima parte di questo lavoro. Negli ultimi cento anni sono state catalogate molte centinaia di molecole e sono state stabilite con precisione le loro costituzioni atomiche. Sono state inoltre trovate molte leggi empiriche riguardanti i tipi di molecole che certi atomi possono entrare a formare.
2 – Le prime idee sulle forze chimiche. Van der Waals
La prima volta che venne avanzata l’ipotesi che le forze che tengono uniti gli atomi tra loro per formare le molecole (le forze chimiche) sono di natura elettrica, fu agli inizi del diciannovesimo secolo.
I primi indizi di questa idea si ebbero proprio nel 1800 quando Nicholson e Carlisle riuscirono a decomporre l’acqua in idrogeno ed ossigeno mediante il passaggio attraverso essa di una corrente elettrica (il fenomeno è quello noto come elettrolisi su cui si iniziò a lavorare, subito dopo la realizzazione della pila da parte di Volta nel 1799). “Se una corrente elettrica riesce a decomporre delle molecole, l’elettricità deve essere alla base delle forze che tengono legati gli atomi costituenti le molecole stesse“: questa era l’idea al suo nascere.
In questo senso si mosse anche, nel 1807, Sir Humphry Davy, il quale, cercando di interpretare alcuni fatti sperimentali (la separazione del sodio dalla soda caustica e del potassio dalla potassa caustica) avanzò l’ipotesi che le forze che legano gli atomi nelle combinazioni chimiche sono di natura elettrica.
Nella sua teoria elettrochimica Davy sostenne le seguenti idee:
1) II contatto fra le particelle, capaci di combinarsi chimicamente, produce la loro elettrizzazione (per contatto queste particelle assumono cariche elettriche di segno opposto).
2) II fatto che particelle si combinino chimicamente consiste in un pareggiamento delle cariche elettriche opposte; il legame chimico si stabilisce tanto più rapidamente quanto più grande è la differenza tra queste cariche.
3) Se una corrente elettrica attraversa un composto, i componenti si separano riacquistando la loro polarità elettrica (negli esperimenti di elettrolisi le cariche positive vanno sull’elettrodo negativo e le cariche negative vanno sull’elettrodo positivo) .
4) Gli elementi hanno maggiore affinità chimica (disponibilità a combinarsi) se hanno maggiore polarità elettrica e viceversa.
5) I processi chimici sono sempre in stretta relazione con i processi elettrici.
Anche Berzelius (1812) si convinse della fondatezza di questa teoria e la sviluppò introducendo nuovi concetti nella chimica. Egli ipotizzò che gli atomi fossero costituiti da una carica positiva e da una negativa (teoria dualistica). Chiamò elettropositivi gli atomi con un eccesso di carica positiva e elettronegativi quelli con un eccesso di carica negativa.
Questa teoria non riusciva però a spiegare alcuni fatti molto importanti (tra cui le leggi dell’elettrolisi di Faraday e alcune questioni inerenti la chimica organica), venne quindi abbandonata per molti anni, fino a quando, nel 1881, Helmholtz non la riprese in un suo lavoro.
Nel frattempo, nel 1873, si stava laureando all’università di Leida in Olanda il non troppo giovane fisico J. D. Van der Waals (era nato nel 1837). Il suo lavoro di tesi riguardava la continuità dello stato liquido e gassoso (e fu pubblicato poi, tradotto in tedesco, a Lipsia nel 1881). Cerchiamo di vedere in un certo dettaglio il lavoro di Van der Waals, tentando di apprezzarne tutta la portata per i futuri sviluppi della fisica.
Sappiamo già che la legge (1660) di Boyle-Mariotte [a temperatura costante la pressione P esercitata su di un gas è inversamente proporzionale al volume V occupato dal gas: PV = K] , la legge (1787) di Charles [se si riscalda un gas, a volume costante, la sua pressione aumenta secondo la relazione: Pt = P0 (1 + αt), dove α (1)è una costante] e la legge (1804) di Gay Lussac [se si riscalda un gas, a pressione costante, il suo volume aumenta secondo la relazione: V = V0(1 + αt), dove α è una costante] che trovano una loro espressione comune nella formula:
pV = p0V0 (1 + αt) => pV = nRT(3) (equazione di stato dei gas perfetti)
si deducono assai semplicemente (come per la legge di Boyle abbiamo già visto) dall’ipotesi di Bernouilli (1797) sulla costituzione dei gas. Per arrivare però a questo risultato si fa una sensibile approssimazione considerando i gas come perfetti, tali cioè che:
1) non esistano forze di attrazione tra le molecole che compongono i gas;
2) le molecole del gas possono essere rappresentate da punti senza dimensioni (abbiamo, cioè, dimensioni nulle e quindi volume nullo).
Ma in realtà non esistono gas perfetti, tutti i gas sono reali: le molecole del gas esercitano tra di loro una debole forza di attrazione ed inoltre queste molecole hanno delle dimensioni diverse da zero, hanno cioè volume. E quando , ad esempio, facciamo il conto che fece Bernouilli nel 1797 (vedi Parte 1) [considerando una massa gassosa contenuta in un recipiente cubico e ammettendo implicitamente che il cammino percorso da una particella che si sposta perpendicolarmente da una parete del recipiente a quella opposta è strettamente uguale alla distanza che separa le due pareti] non teniamo in considerazione il volume della particella.(3) Infatti, per fare un conto più preciso, dovremmo sottrarre alla distanza che separa le due pareti del recipiente il diametro della particella (supposta sferica):

La linea continua corrisponde al percorso effettuato da una particella
secondo il conto fatto da Bernouilli; la linea tratteggiata corrisponde, invece,
al percorso effettivo della particella, quando si tiene conto del suo volume.
Van der Waals ha modificato l’equazione, che abbiamo vista più su, tenendo conto delle correzioni alle quali abbiamo accennato, ed ha ottenuto in sua vece l’equazione:
(p + a/V2)(V – b) = nRT (4)
Nell’espressione che abbiamo scritto la quantità b è un valore costante per ogni tipo di gas e risulta misurabile sperimentalmente, a è anch’esso costante e misurabile sperimentalmente.
Cerchiamo di interpretare da un punto di vista microscopico il significato dell’equazione di Van der Waals.
Le molecole hanno un volume proprio. La somma b dei volumi del le singole molecole costituenti il gas deve essere sottratta al volume V del gas: e questo perché si possa ottenere l’effettivo volume (V – b) che le molecole del gas hanno a disposizione.(5) Inoltre le molecole esercitano, l’una sull’altra, una debole attrazione e questo fatto aumenta di una quantità p’ = a/V2 la pressione che si esercita sul gas: infatti le molecole che si trovano sulla superficie che delimita il volume del gas sono attratte dalle molecole che si trovano più internamente e ciò origina una forza diretta verso l’interno.(6) Vediamo meglio questo fatto.
Supponiamo di avere una molecola all’interno del volume occupato dal gas. Su di essa si eserciteranno delle forze (sulla cui natura e sulla cui legge di variazione con la distanza non facciamo per ora alcuna ipotesi) da parte delle molecole vicine. La risultante di queste forze, su questa molecola interna, è però nulla, in quanto in media questa molecola sarà circondata simmetricamente da un certo numero di altre molecole:
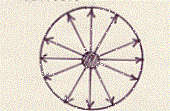
La molecola non sarà allora in alcun modo influenzata da quelle vicine.
Supponiamo ora di avere una molecola vicina alla superficie che delimita il volume occupato dal gas. Le molecole che la circondano, e che esercitano forze su di essa, non saranno più disposte simmetricamente intorno alla molecola stessa:
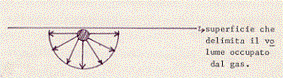
Le forze non si faranno allora più equilibrio ed avranno una risultante, non nulla, diretta verso l’interno. Questa forza risultante agisce sulla molecola in modo tale da rallentarne l’urto contro la parete del recipiente.
Ricordando che sono proprio gli urti delle particelle costituenti un gas, contro le pareti del contenitore, che originano la pressione, si capisce subito che, diminuendo l’intensità di questi urti, si diminuisce la pressione esercitata dal gas.
E proprio questo è il significato del termine a/V2, introdotto nell’equazione di Van der Waals.
Si possono allora cominciare a trarre delle prime conclusioni:
il termine p’ = a/V2 è responsabile delle forze attrattive tra le molecole, mentre il termine b è in qualche modo responsabile della repulsione tra le molecole (il volume proprio occupato dalle molecole fa si che queste al massimo possono essere avvicinate fino a toccarsi: impenetrabilità della materia). (7)
Abbiamo riportato con maggiori dettagli l’opera di Van der Waals perché essa fu gravida di conseguenze. Grazie alla sua equazione (ed alle ipotesi da lui fatte relative alla pressione interna ed al covolume) da quel momento le deboli forze di attrazione che si esercitano tra le molecole a breve distanza e le notevoli forze di repulsione che si esercitano tra le stesse a brevissima distanza presero il suo nome (forze di Van der Waals).
In quegli anni nulla si sapeva sulla natura di queste forze, ol tre all’ipotesi, che si faceva, che fossero di natura elettrostatica.(8) Ma da allora, appunto, le azioni che si esercitano tra le molecole in un aggregato qualsiasi, e che abbiamo citato qualche riga più su, sono chiamate forze di Van der Waals.
Ancora oggi la natura di queste forze non è completamente chiara, anche se si sono fatti dei buoni passi avanti.
Tra qualche pagina affronteremo una discussione più dettagliata di queste forze inserendole nel contesto storico in cui cominciarono ad avere una spiegazione.
Intorno al 1885 gli studi sull’elettrolisi si erano arricchiti di una notevole mole di esperienze ed Arrhenius in una sua teoria sulla dissociazione elettrolitica avanzò l’ipotesi secondo cui i sali diluiti in soluzioni acquose sono dissociati in ioni positivi e negativi. (Questa teoria venne adottata ed ampliata successivamente, nel 1891, da Werner il quale introdusse nel problema i concetti di valenza principale e di valenza ausiliaria).
Un notevole impulso alle teorie che volevano di natura elettrica il legame tra atomi nella formazione delle molecole venne dopo il 1897 quando J.J.Thomson scoprì l’elettrone misurandone la carica e la massa con una certa precisione. Il fatto che l’elettrone fosse un costituente comune di tutti gli atomi aprì nuove entusiasmanti prospettive.
Lo stesso Thomson sviluppò (1907) una teoria del legame chimico(10) basata appunto sul fatto che gli elettroni dovevano avere una parte preponderante nella formazione dei legami tra atomi. Thomson sosteneva che vi sono degli atomi elettropositivi e degli atomi elettronegativi. Sono elettropositivi quegli atomi che perdendo degli elettroni riescono a raggiungere una configurazione stabile; elettronegativi quelli che raggiungono questa stabilità di configurazione acquistando degli elettroni degli atomi vicini.
Su questa strada si mossero, all’incirca nello stesso periodo: Abegg (1904), Ramsay (1908), Falk e Nelson (1910 ÷1918), Bray e Branch (1915) ed altri chimici e fisici.
Intorno al 1914 si ebbe poi ancora un contributo di J.J.Thomson il quale studiando vari tipi di molecole le distinse in molecole polari(10) (quelle del tipo NaCl = cloruro di sodio = sale da cucina) e molecole non polari (quasi tutte quelle costituenti le sostanze organiche). Inoltre precisò alcuni concetti collegati alla sua precedente teoria del legame chimico: la valenza elettropositiva dell’atomo in un elemento è uguale al numero di elettroni che questo atomo può perdere facilmente, la valenza elettronegativa è invece uguale alla differenza tra il numero otto e la valenza elettopositiva.
Un notevole passo avanti, nella soluzione del problema del legame chimico, fu fatto dopo che Bohr nel 1913 propose il suo modello atomico.
Nel 1916 i due fisici W.Kossel (tedesco) e G.N.Lewis (statunitense) svilupparono una teoria che oggi è conosciuta come teoria elettronica del legame chimico. In particolare W.Kossel, studiando le molecole polari (quelle ionizzabili, quelle cioè a cui possono essere tolti elettroni con facilità), elaborò la teoria del legame ionico; G.N.Lewis, per tener conto della formazione delle molecole non polari, sviluppò invece una seconda teoria (notevolmente estesa da I. Langmuir nel 1919), quella del legame covalente.
Va detto comunque che la natura delle forze che entrano in gioco in questi legami rimane oscura. Bisognerà aspettare la meccanica quantistica (dal 1926 in poi) per avere una soluzione di que sto problema.
Vediamo un poco più da vicino questi due tipi di legame tra atomi, ricapitolando prima un poco sull’atomo di Bohr e sul principio di Pauli.
3 – Legame ionico e legame covalente
Come abbiamo già detto il modello di Bohr, a cui applichiamo il principio di Pauli (su un livello energetico vi possono essere al massimo due elettroni se hanno spin opposti), raccoglie in gruppi tutte le orbite che possono essere occupate da elettroni (orbite permesse), ciascuno dei quali viene ad occupare uno strato nello spazio che circonda il nucleo dell’atomo. Questi strati possono contenere un numero limitato di elettroni; per esempio lo strato più interno, formato da un solo livello energetico (1 s) può contenere al massimo due elettroni; lo strato successivo, formato da quattro livelli energetici (2s, 2px, 2py, 2pz), può contenere al massimo otto elettroni. Nella tabella seguente sono riportati da un lato il diagramma dei valori delle energie dei livelli energetici – elettronici che si hanno intorno al nucleo [le riquadrature indicano i vari gruppi di livelli che formano i successivi strati] , dall’altro il numero degli elettroni che al massimo si possono trovare in ogni strato:
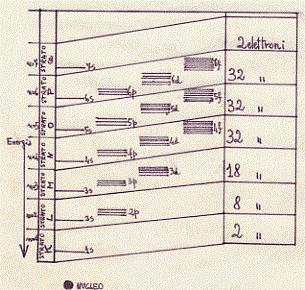
In questo schema ci interessa solo l’ordine di successione dei livelli dal basso in alto,
e non il valore numerico della loro energia che non potrebbe essere precisato se non
atomo per atomo. Il disegno non è fatto in scala e può valere per molti elementi.
La successione degli elementi atomici nella tavola periodica presenta il riempimento successivo di questi strati da parte degli elettroni.
Vediamo qualche esempio.
Il primo elemento della tavola periodica è l’idrogeno che ha un solo elettrone il quale si troverà sul livello 1s nello strato K. Il secondo, l’elio, ha due elettroni che si troveranno sul livello 1s nello strato K (il quale risulterà completo). Il terzo, il litio, ha tre elet troni, due dei quali si troveranno sullo strato K riempiendolo, mentre il terzo si troverà nel livello 2s dello strato L.
Si va avanti così fino al neon che ha 10 elettroni due dei quali completano lo strato K, i rimanenti otto, invece, completano lo strato L. Con il sodio, 11 elettroni, completati gli strati K ed L, un elettrone andrà ad occupare un livello energetico dello strato M.
Si va così avanti via via riempiendo i livelli energetici più vicini al nucleo.
Con il raffinarsi del modello atomico descritto, si trovò il modo di usarlo per spiegare le regole chimiche della valenza.
Il fatto che i gas nobili non entrano con facilità in legami chi mici con altri elementi attirò l’attenzione di Kossel sul fatto che gli atomi sono particolarmente stabili quando i loro strati elettronici sono completi, così che, per raggiungere questa stabilità, un atomo cercherà di acquistare o perdere elettroni.
Gli elettroni che si trovano nell’ultimo strato dell’atomo sono gli elettroni di valenza.
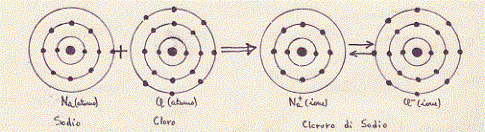
Nel caso del doro (Cl), lo strato più esterno è mancante di un elettrone per essere completo. Il cloro quindi, per la sua stabilità, cercherà di unirsi con un atomo che nel suo strato esterno ha un solo elettrone. II sodio (Na) ha un solo elettrone nel suo strato più esterno e tenderà quindi a perderlo con facilità per rimanere con lo strato immediatamente più interno completamente occupato. Il cloro ed il sodio si uniranno quindi con facilità originando il cloruro di sodio (NaCl o sale da cucina). Il cloro ed il sodio si legano quindi con un legame particolare: quello ionico.
4 – Legame ionico (Kossel).
La teoria di Kossel del legame ionico spiega la formazione degli ioni e l’unione di ioni positivi (atomi a cui mancano elettroni) e negativi (atomi che hanno degli elettroni in più) mediante la forza di attrazione coulombiana.(11)
Nel legame ionico si ammette il passaggio di elettroni da un atomo ad un altro.
Supponiamo di avere un elemento (ad es. il sodio Na) che abbia sull’ultimo strato un numero di elettroni inferiore a quattro ed un al tro elemento (ad es. il doro Cl) che abbia sull’ultimo strato un numero di elettroni superiore a quattro. Poiché la configurazione più stabile è quella in cui un atomo ha il suo ultimo strato completo di elettroni e poiché quest’ultimo strato è generalmente completo quando ha otto elettroni, è evidente che l’atomo con un numero di elettroni inferiore a quattro sull’ultimo strato tenderà a perderli, mentre l’atomo che ha un numero di elettroni superiore a quattro nell’ultimo strato tenderà ad acquistarne in numero tale da far si che quest’ultimo strato risulti completo (cioè con otto elettroni). Questa configurazione risulterà molto stabile (si sarà formata una molecola di NaCl).
Vediamo più in dettaglio, come esempio, il caso della molecola, più volte citata, di cloruro di sodio (NaCI).
Nella formazione del cloruro di sodio, il metallo (Na) che, come tale, possiede meno di quattro elettroni di valenza (uno soltanto), si ionizza, perde cioè l’unico elettrone che possedeva nell’ultimo strato, acquistando di conseguenza una carica positiva (Na+); il non metallo (Cl) che ha nell’ultimo guscio sette elettroni di valenza, assume l’elettrone perduto dal metallo (Na) e diviene anch’esso uno ione, dotato però di una carica negativa (Cl–). Fra i due ioni, che possiedono entrambi una carica elettrica, ma di segno opposto, si esercita una forza elettrostatica (la forza di Coulomb) che li tiene uniti, formando un legame tra ioni, un legame, cioè, ionico. In questo processo, naturalmente, è necessario che il numero degli elettroni perduto da una specie atomica sia uguale a quello acquistato dall’altra specie (è necessario quindi un atomo di sodio ogni atomo di cloro) .
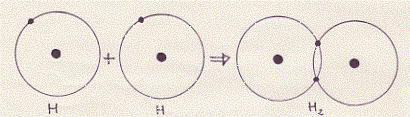
Un altro tipo di legame, per mezzo degli elettroni (e quindi originato da una forza di tipo elettrico), tra gli atomi si realizza, secondo la teoria di Lewis e Langmuir, nell’unione di due o più atomi nella formazione di un composto non polare(12) (come ad esempio nelle molecole più semplici: H2 , Cl2 , O2 ). In questo caso, poiché non si formano ioni, il legame è detto non polare (o covalente).
5 – Legame covalente (Lewis-Langmuir)(13)
Nella molecola composta (ad esempio) da atomi di uno stesso elemento, i singoli atomi, uguali fra loro, per raggiungere il riempimento completo dell’ultimo strato, mettono in comune (appunto in questo ultimo strato) uno o più elettroni.
Secondo la spiegazione data parlando del principio di Pauli, gli atomi dovrebbero tendere a respingersi, poiché, quando essi si avvicinavano, gli elettroni, per evitare di occupare stati già occupati, devono portarsi in stati ad energia più elevata; tenendo conto però che alcuni livelli energetici (meglio: alcuni strati) sono incompleti, il legame si forma attraverso questi livelli energetici. Quando gli strati più esterni degli atomi sono completi, l’eventuale legame dovrebbe avvenire mediante livelli di energia superiore a quelli degli strati completi; se, però, l’energia necessaria per portare gli elettroni a livelli ener getici superiori è considerevole, la repulsione tra gli atomi impedisce la formazione del possibile legame; è questo il caso degli strati completi dei gas nobili (o inerti) a cui abbiamo già accennato.(14)
6 – Ritorniamo a Van der Waals. Legame tra molecole.
Come avevamo accennato, parlando di Van der Waals, la natura delle forze, che si esercitavano tra molecole originando la loro attrazione (a/V2) e la loro repulsione (b) non era affatto chiara. Si cer cò quindi per diversi anni di spiegare la natura delle forze di Van der Waals.
Nel 1885 Boltzmann, riprendendo un lavoro di J.C.Maxwell (1868),(15) iniziò ad affrontare il problema dell’esatta natura di queste forze, studiando il loro campo di azione e la loro dipendenza dalla distanza tra le molecole. Il lavoro di Boltzmann non portò degli utili risultati; egli costruì comunque un modello in termini di energia (una buca di potenziale in tre dimensioni con barriere verticali) che, secondo lui, avrebbe spiegato i fatti sperimentali fino ad allora conosciuti.
Nel 1886 cominciò a studiare il problema W.Sutherland, il quale provò ad applicare ai molti dati sperimentali che aveva a disposizione una legge che fornisce la forza F di attrazione tra due molecole di massa m 1 ed m2 ad una distanza r:(16)
F = A.(m1m2)/r4
(dove A è un parametro che varia da sostanza a sostanza) questa legge gli era stata suggerita da precedenti lavori di Laplace (Teoria generale dell’azione capillare – 1806) e di Gauss (Principia Generalia – 1830). Sutherland, operando in questo modo, riuscì a spiegare alcuni fatti sperimentali (ad esempio: l’azione capillare e l’effetto Joule-Thomson (leggi: Kelvin) relativo ad un gas fortemente compresso che attraversa una parete porosa raffreddandosi: effetto che dimostra che la coesione tra i gas non è nulla).
Qualche anno dopo, nel 1893, lo stesso Sutherland ipotizzò qual cosa di più interessante per gli sviluppi futuri. Egli stabilì una stretta e generale correlazione tra il parametro A della sua formula con la polarizzabilità(17) degli atomi delle sostanze che agiscono per formare le molecole, e, di conseguenza, provò a studiare le forze interatomiche ed intermolecolari tenendo conto dell’intrinseca struttura degli atomi e delle molecole interessati ai fenomeni.
Ancora Sutherland, nel 1902, sviluppò ed ampliò le precedenti idee. Intanto, nello stesso periodo (tra il 1901 ed il 1903), anche il fisico M.Reniganum si stava interessando al problema. Sutherland ed, indipendentemente, Reniganum ipotizzarono che le molecole neutre trasportano delle cariche elettriche positive e negative che sono concentrate interamente o parzialmente in due punti. Ragionando in questi termini, per la forza di attrazione tra le molecole, si trova che questa forza varia con la distanza tra le molecole come 1/r4 . Niente però dava ragione a questo andamento. Molti altri fenomeni erano bene spiegati da leggi empiriche con andamenti 1/r5 e 1/r6 (questi fenomeni erano stati studiati, mediante l’applicazione di queste leggi empiriche, sempre da Sutherland tra il 1886 ed il 1893).
Con lo stesso metodo di Sutherland (utilizzando cioè una legge di forza, scelta allo scopo, ed andando ad analizzare poi i dati sperimentali al variare di alcuni parametri che compaiono nella legge) lavorarono più tardi, dal 1924 in poi, anche J. E. Lennard e Jones ed i loro assistenti.
I due fisici usarono una legge in cui erano considerate le forze attrattive e quelle repulsive oltre a dei parametri, modificando i quali dal confronto con i dati sperimentali, si arrivava ad un aggiustamento dell’intera legge.
La legge da loro usata era della forma:
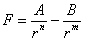
dove il primo termine rappresenta la forza repulsiva; il secondo la forza attrattiva; A, B, n ed m (con n > m)(18) sono i parametri da modificare via via; r è la distanza tra gli atomi o le molecole che si considerano; F è la forza risultante.
Anche per Lennard e Jones le costanti A e B, diverse per diverse sostanze, erano in qualche modo legate alla struttura intrinseca degli atomi e delle molecole (in particolare alla loro polarizzabilità).
Con questo metodo si sono ottenuti dei buoni risultati.
7 – Diagrammi di energie e forze relativi a molecole
Vediamo ora meglio, aiutandoci con un grafico, il significato di una legge di tipo or ora visto.
Supponiamo di avere tanti atomi (o molecole) in moto rapido e casuale (è quanto si verifica, ad esempio, in un gas). L’energia del moto è l’energia cinetica mentre la temperatura è la misura dell’energia cinetica di questi atomi (o molecole).
Quando due atomi (o molecole), si avvicinano tra di loro considerevolmente rispetto agli altri atomi (o molecole), allora c’è la possibilità che si formi un legame chimico. Supponiamo che questi due atomi (fissiamo le idee su due atomi) si possano rappresentare come due sfere rigide,(19) vediamo cosa succede, in termini di energia (e di forza),(19) quando questi due atomi si avvicinano tra di loro fino ad arrivare a contatto. Costruiamoci dei grafici:
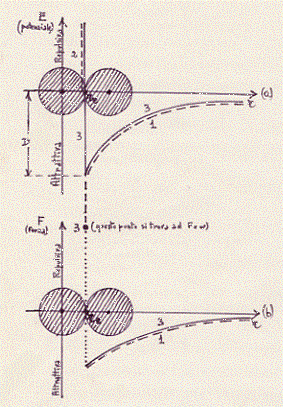
Nella figura: r rappresenta la distanza fra i centri degli atomi.
Le curve tratteggiate ed indicate con 1 danno la parte attrattiva.
La curva tratteggiata ed indicata con 2 dà la parte repulsiva.
Il punto indicato con 3 dà la parte repulsiva (questo punto si
trova ad una F = ∞). Le curve a tratto continuo (compreso il
punto di cui prima) indicate con 3 danno l’effetto risultante.
[Riguardo al punto indicato con 3 c’è da dire che, al momento
del contatto tra i due atomi, c’è una forza di repulsione infinita e
questa forza infinita è rappresentata, appunto dal punto indicato
con 3J. La re è di equilibrio. Intorno a questo valore di r la molecola
compie delle piccole oscillazioni di origine termica. La quantità
D riportata in figura rappresenta l’energia necessaria per
dissociare la molecola: questa energia è tanto più piccola quanto più
piccolo è il valore di D. Più è grande D più la molecola è stabile.
I grafici di figura rappresentano la seguente situazione: a grandi distanze(20) i due atomi esercitano una debole attrazione l’uno sull’altro; questa forza di,.attrazione diminuisce molto rapidamente con l’aumenta re della distanza;(21) se gli atomi si avvicinano la forza di attrazione cresce e raggiunge il valore massimo quando le due sfere rigide si toccano; un tentativo di avvicinamento ulteriore comporta una violenta (infinita) repulsione (completa impenetrabilità della materia).(22) Quando due atomi entrano nella composizione di una molecola si stabiliscono nella buca di energia potenziale che appare disegnata in figura,compiendo delle piccole oscillazioni, di origine termica, intorno alla posizione (re ) di equilibrio.(23)
Consideriamo ora questi due atomi che si avvicinano, secondo il modello atomico di Bohr-Pauli (modello atomico di Bohr con l’introduzione del principio di Pauli).
In questo caso, in termini di forza e di energia, si hanno i grafici seguenti:
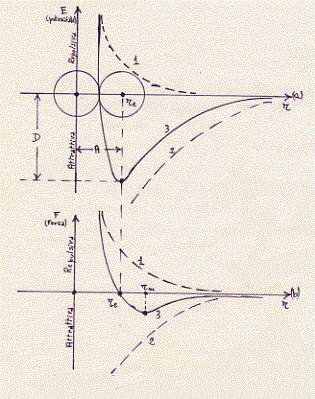
(a)- Curva dell’energia potenziale di una molecola biatomica in
funzione della distanza r tra i nuclei degli atomi che la costituiscono.
La curva 1 è relativa alla repulsione; la curva 2 all’attrazione;
la curva 3 è quella risultante. Il punto di ascissa re è quello di
equilibrio:la molecola che si forma vibra intorno a questa posizione.
(b)-Curva della forza che si esercita tra due atomi costituenti una
molecola biatomica in funzione della distanza r tra i nuclei.
La curva 1 è relativa alla repulsione; la curva 2 all’attrazione;
la curva 3 è quella risultante. II punto di ascissa rm è quello
di massima forza attrattiva tra le molecole.
I grafici di figura precedente rappresentano la seguente situazione: a grandi distanze i due atomi esercitano una debole attrazione l’uno sull’altro; questa forza di attrazione diminuisce molto rapidamente con l’aumentare della distanza; se gli atomi si avvicinano la forza di attrazione cresce e raggiunge il valor massimo per un dato valore (rm), sempre piccolissimo, della distanza fra i nuclei atomici; un avvicinamento ulteriore comporta una diminuzione dell’attrazione ed infine, ad una distanza A (sull’ascissa di figura, chiamata distanza di equilibrio), ten de a zero. Quando due atomi entrano nella composizione di una molecola si stabiliscono nella buca di energia potenziale che appare disegna ta in figura, compiendo piccole oscillazioni, di origine termica, intorno alla posizione di equilibrio. Per una distanza tra gli atomi minore di A si ha il sorgere di forze di repulsione che crescono molto rapidamente e rendono praticamente impossibile l’avvicinamento ulteriore dei due atomi.[Forze molto simili a quelle agenti tra gli atomi agiscono tra molecole. Le caratteristiche essenziali dell’attrazione tra molecole sono ancora rappresentate dalla curva dell’energia potenziale e dalla curva della forza tracciata per gli atomi (va tuttavia osservato che tra i due tipi di attrazione vi sono differenze sostanziali come risulterà implicitamente da quanto diremo oltre].
Confrontiamo in un dettaglio le due ultime figure . Se gli atomi fossero, come avevamo ipotizzato per la costruzione dei grafici della prima, delle sfere rigide, la forza di repulsione che si eserciterebbe tra di essi sarebbe nulla nel caso in cui la distanza tra i due atomi fosse maggiore della somma dei raggi delle due sfere; questa forza sarebbe in vece infinita nel caso in cui la distanza tra i due atomi fosse minore della somma dei raggi delle due sfere. Nell’ultima figura abbiamo invece visto che, con l’introduzione dell’atomo di Bohr-Pauli, la curva risultante ha un andamento continuo e regolare. Questo fatto suggerisce che gli atomi non sono completamente impenetrabili. In ogni caso l’impenetrabilità della materia, che si manifesta nella piccola compressibilità dei solidi e dei liquidi, può essere considerata come conseguenza del principio di Pauli che origina, appunto, le notevoli forze repulsive ad una piccolissima distanza tra gli atomi costituenti la molecola. Infatti, se due nuclei appartenenti a due atomi potessero avvicinarsi l’uno all’altro mentre gli elettroni intorno ad essi rimanessero indisturbati, alla fine i nuclei (a parte la repulsione elettrostatica) si toccherebbero, formando un unico nucleo, e più elettroni, provenienti da atomi diversi, si verrebbero a trovare nella stessa orbita. La repulsione tra gli atomi dipende allora dal fatto che gli elettroni dovrebbero sistemarsi su orbite diverse (a causa del principio di Pauli) acquistando in definitiva energia. Questo acquisto di energia da parte degli elettroni è grande ed allora gli atomi si respingono (ci vuole meno energia per respingerli che non per far acquistare energia agli elettroni). Oltre alla repulsione dovuta al principio di Pauli c’è una piccola repulsione elettrostatica.(24)
Vediamo di confrontare con un grafico analogo ai precedenti i valori delle attrazioni e repulsioni elettrostatiche delle repulsioni dovute al principio di Pauli, delle curve teoriche e sperimentali:
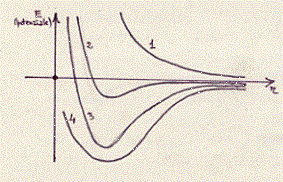
La curva 1 è relativa alla repulsione dovuta al principio di Pauli;
la curva 2 è relativa all’attrazione e repulsione elettrostatica;
la curva 3 è quella ricavata teoricamente (25) e comprende la
repulsione di Pauli e l’attrazione e repulsione coulombiana;(26)
la curva 4 è quella ricavata da dati sperimentali. (27)
[Si osservi che il piccolo minimo della curva 2 non spiegherebbe la
stabilità delle molecole] .
In breve prima di ritornare a dove avevamo lasciato il discorso, ricapitoliamo brevemente questi ultimi concetti (aggiungendone un paio) dicendo che:
1) Gli atomi di una molecola interagiscono a causa di due tipi di forze di natura essenzialmente elettrica (almeno in origine).
2) In primo luogo vi sono forze attrattive che interessano il legame chimico (forze di valenza). Questo tipo di forze decresce molto rapidamente al crescere della distanza tra i due atomi.
3) In secondo luogo vi sono forze repulsive trascurabili a grande distanza ma che crescono più rapidamente di quelle attrattive a piccolissima distanza (impenetrabilità della materia).
4) Ad una certa distanza tra gli atomi (punto di ascissa re della penultima figura) si ha una situazione di equilibrio e gli atomi possono rimanere a tale distanza per un tempo indefinito [e questa è la distanza che normalmente separa gli atomi in una molecola o in un aggregato qualsiasi (ad es.: un solido)].
5) Gli atomi vibrano intorno a questa posizione di equilibrio e l’ampiezza dell’oscillazione è proporzionale all’aumento di temperatura (moti di vibrazione).
6) Inoltre una molecola può ruotare su se stessa con una velocità (angolare) che cresce anch’essa con la temperatura (moti di rotazione).
7) Infine tutta la molecola può muoversi lungo una determinata direzione con un moto di traslazione (moto di traslazione).
***
Prima di tornare ancora sulle forze di Van der Waals, per cercarne le successive spiegazioni, occupiamoci per un momento della definizione di alcune grandezze e concetti che abbiamo già incontrato e che ci serviranno in seguito: dipolo elettrico, dipolo indotto, polarizzabilità, orientamento dei dipoli sotto l’azione di un campo elettrico, molecole polari e dipolo istantaneo.
NOTE
(1) La costante a rappresenta l’aumento di energia delle molecole per ogni grado centigrado di aumento di temperatura α = 1/273. Ma si sta parlando di pressione e volume, che c’entra l’energia? Vediamolo. Ricordiamo che l’espressione più generale che conosciamo per l’energia è E = mv2 , che si può scrivere: E = m(l2/t2 ). Troviamoci le dimensioni di questa energia: [E] = [M] [L]2 [T]-2.
Calcoliamoci ora le dimensioni del prodotto pV di una pressione per un volume (che incontreremo tra qualche riga) che si può anche scrivere:
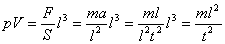
Si ha:
[pV] = [M][L]2[T]-2 = [E]
Come si può quindi vedere il prodotto pressione per volume ha le dimensioni di una energia!
(2) Dove n è un numero di grammomolecole, R è la costante universale dei gas e T è la temperatura assoluta o temperature Kelvin [T °K = (t+273)°C].. Cerchiamo di vedere come si passa dalla prima alla seconda relazione scritta. Introducendo nella prima equazione la temperatura assoluta T = t + 273 => t = T – 273,
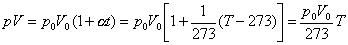
Ricordando ora la legge di Avogadro [alla temperatura t0 = 0°C ed alla pressione p0= 1 atmosfera, cioè a T0 = 273°K, ogni grammomolecola di gas occupa un volume di litri 22,4. Se avremo quindi n grammomolecole avremo un volume V0 = 22,4 x n ( si può scrivere:
pV = (22,4/273).nT
Indicando poi con R la costante universale dei gasi [ R = 22,4/273 (litri.atmosfere/gradi.grammomolecole) = 0,821 (litri.atmosfere/gradi.grammomolecole) si ha la relazione che cercavamo: pV = nRT.
(3) Osserviamo a parte che la legge di Boyle (pV = K), se non sottoposta a limiti di validità, condurrebbe ad un assurdo. Infatti, secondo questa legge, se si aumenta la pressione P infinitamente il volume V di una determinata massa di gas si dovrebbe ridurre a zero.
(4) Ha cioè aggiunto al termine p, che compare nell’equazione dei gas perfetti, il termine a/V2 ed al termine V, che compare sempre nell’equazione dei gas perfetti, il termine -b.
(5) La costante b è comunemente chiamata “covolume”. Osserviamo a parte che il volume effettivamente occupato dalle molecole di un gas è soltanto un quarto di b (come si può calcolare).
(6) p’ = a/V2 = pressione interna. Si osservi che tale pressione deve essere tanto maggiore quanto più le molecole sono vicine; per questo motivo essa risulta inversamente proporzionale al quadrato del volume V occupato dal gas.
(7) Per capire meglio questo concetto vedi più avanti (anticipiamo che il responsabile di questa repulsione tra molecole, quando si trovano a brevissima distanza, è il principio di Pauli).
(8) Cerchiamo di vedere perché si pensava a forze di natura elettrostatica e non a forze di natura gravitazionale. Ricordiamo che la legge della gravitazione universale di Newton (1666) è:

FN = G.(m1m2/r2)
dove G = 6,67. 10 N.m2/kgm2 è una costante, m1 ed m2 sono le due masse che si attraggono ed r è la distanza tra di esse. Ricordiamo poi che la legge di Coulomb (1785) che studia l’attrazione (o repulsione) elettrostatica tra due cariche elettriche q1 e q2 alla distanza r, è:

FC = K(q1q2/r2)
dove K è una costante che vale: K = 8,98. 109 N.m2/coulomb2. Prendiamo ora in considerazione due elettroni (che sappiamo avere ciascuno una carica qe = 1,60.10-19 coulomb ed una massa me = 9,11.10-31Kgm), supponiamo che siano ad una distanza dell’ordine di grandezza delle dimensioni atomiche (r ~ 10-10 m), applichiamo ad essi le leggi di Newton e di Coulomb e confrontiamo i valori che si ottengono per le rispettive forze. Applicando la legge di Newton si trova:
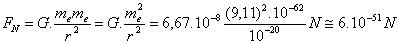
Applicando invece la legge di Coulomb si ha:
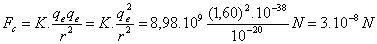
Confrontando le due forze si trova:
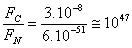
e come si vede la forza di Coulomb, cioè la forza elettrostatica risulta essere 1047 volte maggiore di quella gravitazionale.
(9) Ampliata successivamente (1910) da J.Stark il quale introdusse per primo il concetto di elettroni di valenza per indicare i legami che si hanno tra gli atomi nei composti.
(10)Vedi più avanti.
(11) Se abbiamo due cariche elettriche q1 e q2 , ad una distanza r queste si attraggono (se sono dello stesso segno) o si respingono (se sono di segno opposto) con una forza data dalla legge di Coulomb:
F = K (q1q2/r2)
dove K è una costante di proporzionalità.
(12) Vedi più avanti.
(13) Il legame covalente fu studiato quantisticamente da Heitler e London nel 1927 (vedi più avanti).
(14) Un legame dello stesso tipo di quello covalente si ha tra molecole. Questo legame è chiamato dativo (vedi più avanti).
(15) L’opera più importante di Maxwell, il Trattato di elettricità e magnetisimo, fu pubblicata nel 1873.
(16) Secondo Sutherland le molecole che agiscono sono sempre ad una certa distanza tra loro: r non può mai annullarsi.
(17 )La polarizzabilità misura quanto un campo elettrico distorce un atomo od una molecola, spostando gli elettroni dell’atomo o della molecola rispetto al nucleo positivo (vedi comunque più avanti).
(18) Il fatto che n sia maggiore di m fa si che le forze repulsive abbiano un raggio di azione più piccolo di quello delle forze attrattive. Da notare che la forza repulsiva è positiva mentre quella attrattiva è negativa. I valori più in uso, oggi, per n ed m sono: n = l2 ed m = 7 (Potenziale 12-6). Osserviamo a parte che questo modo di procedere è ancora oggi quello in uso, fornendo una accurata espressione teorica per le forze intermolecolari.
(19) Questa prima approssimazione è quella che in sostanza fece Van der Waals e che venne ripresa nel 1912 da Keesom (vedi più avanti).
(20) Abbiamo già visto la legge di Coulomb. Ricordiamo che Fc è negativa se le due cariche hanno segno opposto e quindi quando c’è attrazione; mentre Fc è positiva se le due cariche hanno stesso segno, quindi quando c’è repulsione. Per comprendere meglio quanto segue, vediamo graficamente qual è la legge dell’attrazione e della repulsione elettrostatica in termini di forza e di energia:

(21) Relativamente alle dimensioni atomiche.
(22) Praticamente ad una distanza di 6 o 7 Å l’energia di legame tra due atomi si annulla. Non si ha più, allora, una molecola, ma si hanno due atomi separati.
(23) Secondo il modello di atomi (o molecole) a sfere rigide la materia risulterebbe completamente impenetrabile. In realtà questo fatto è in contrasto con l’esperienza in quanto, a parte i gas, i liquidi ed i solidi risultano comprimibili in un modo che lascia intendere la non completa penetrabilità della materia.
(24) Mentre la repulsione dovuta al principio di Pauli va come r-6, la repulsione elettrostatica va come r-13.
(25) Le curve teoriche sono state studiate da Heitler e London nel 1927, da Siguira nel 1927, da Born ed Oppenheimer nel 1927, da Wang nel 1928, da Morse nel 1929 (si noti che le curve del tipo da noi disegnato sono oggi conosciute come curve di Morse) e da altri più tardi.
(26) Si può dimostrare che solo il 14% dell’energia totale di legame tra molecole esistenti in natura può essere considerata come elettrostatica. Questo dato è stato ripreso dal testo di Syrkin e Dyatikna – Molecular Structure – pagg. 65 e 66.
(27) Le tecniche sperimentali usate per studiare le molecole sono:
1) Spettroscopia
2) Diffrazione di raggi X
3) Diffrazione di elettroni (o neutroni)
L’interpretazione dei dati sperimentali è possibile solo con l’introduzione della meccanica quantistica.
Categorie:Senza categoria

Rispondi