Dalle prime idee sulla costituzione atomica e sulle forze interatomiche alla struttura dell’atomo e delle molecole secondo la fisica dei quanti.
PARTE 2: MOLECOLE
1 – La teoria dell’Orbitale Molecolare (O.M.): molecole biatomiche omonucleari ed eteronucleari
Un altro metodo approssimato per lo studio quanto-meccanico delle molecole, che per certi versi è analogo al precedente e per altri differente, fu introdotto nello stesso armo (1927) del metodo L.V.: prende il nome di metodo dell’orbitale molecolare (O.M.).
Il primo lavoro sull’argomento fu scritto, appunto nel 1927, dal danese Øyvind Burrau e si occupava soltanto della molecola-ione idrogeno H2+ . Questo metodo fu subito ripreso ma riuscì ad essere completamente sviluppato solo quando il metodo L.V. era già affermato.
Il primo articolo che sviluppava le idee di Burrau fu del fisico-chimico statunitense E.U. Condon (1927) che trattò la molecola H2 di idrogeno. Contributi determinanti, che valsero a strutturare completamente il metodo,vennero negli anni successivi: nel 1928 dallo statunitense R.S. Mulliken e dal tedesco F. Hund; nel 1929 ancora da Mulliken, quindi dal britannico J. E. Lennard-Jones e dal tedesco G. Herzberg; nel 1930 ancora da Hund; nel 1931 e nel 1932 ancora da Mulliken. Tutti i contributi qui elencati sono relativi principalmente a molecole biatomiche; ve ne furono naturalmente anche di relativi a molecole poliatomiche: del tedesco M. Dunkel nel 1930; di Hund nel 1931 e 1932; di Mulliken nel 1932 (in questo anno egli introdusse la terminologia orbitale molecolare) e nel 1933.
Vediamo su quali basi ed ipotesi prende le mosse il metodo O.M. avvertendo subito che molto spesso esso risulta più semplice e descrittivo del metodo L.V., anche perché è praticamente un’estensione alle molecole degli orbitali atomici.
Secondo il metodo O.M. si considerano i nuclei degli atomi che vanno a fermare la molecola già situati nelle loro posizioni di equilibrio nella molecola stabile e si vanno quindi a studiare le funzioni d’onda y degli elettroni, tenendo conto che si passa da funzioni d’onda atomiche a funzioni d’onda molecolari. Non si considerano più gli atomi come un tutt’uno che va ad interagire al momento di formare la molecola, ma si discute di cerne sono gli orbitali per elettroni che non sono più associati ad un solo nucleo ma a due o più (supposto, come abbiamo detto, che questi nuclei siano già nella loro posizione di equilibrio molecolare).
Tenendo conto che vale anche qui l’approssimazione del campo “self-consistent” (lo si vada a rileggere), descriviamo i criteri con cui gli elettroni debbono essere inseriti nella molecola:
1) a ciascun elettrone, nella molecola, è associata una funzione d’onda y detta orbitale molecolare; questa funzione d’onda ha lo stesso significato della funzione d’onda y atomica;
2) gli orbitali molecolari sono policentrici, sono associati cioè a due o più nuclei;
3) ad ogni orbitale molecolare sono associati tre numeri quantici n, l, l (che sostituisce il quanto magnetico m che si aveva nel caso di orbitali atomici)(1) i quali ne definiscono l’energia e la forma; inoltre ad ogni elettrone in un dato orbitale molecolare è associato il quarto numero quantico, quello di spin ms, che può assumere i due valori ± 1/2 (sempre in unità h/2p);
4) ad ogni orbitale molecolare compete una determinata energia; la somma delle energie dei singoli elettroni che occupano i vari orbitali molecolari ci fornisce l’energia complessiva della molecola (si debbono però fare le correzioni relative alle interazioni tra gli elettroni);
5) possiamo sistemare gli elettroni sui vari orbitali permessi uno alla volta secondo il principio di aufbau (si vada a rileggere) e tenendo conto che vale il principio di Pauli (su ogni orbitale molecolare possono trovare posto al massimo due elettroni e ve ne sono due se questi hanno spin opposti).
Tra i vari modi di ottenere gli orbitali molecolari noi sceglieremo quello più in uso: il metodo della combinazione lineare degli orbitali atomici (L.C.A.O.).
2 – L’approssimazione L.C.A.O.
La discussione che faremo ora sarà riferita alle molecole biatomiche. Più avanti vedremo le poliatomiche.
Disponiamo quindi di due nuclei atomici vicini (chiamiamoli A e B) in posizione stabile e formanti (insieme ad. alcuni elettroni) la molecola in considerazione. Consideriamo un elettrone intorno a questi due nuclei e vediamone il comportamento da un punto di vista classico. Trascurando la repulsione tra i nuclei, che verrà presa in considerazione in seguito, questo elettrone si muoverà in una orbita che ha come centri i due nuclei. Nel suo movimento l’elettrone passerà alternativamente più vicino al nucleo A e più vicino al nucleo B. A queste punto si fa una approssimazione: quando l’elettrone si trova nelle vicinanze del nucleo A si assume che sia influenzato solo da A e dagli elettroni vicini ad A ed, analogamente, quando l’elettrone si trova nelle vicinanze del nucleo B si assume che sia influenzato solo da B e dagli elettroni vicini a B (questo fatto è chiaramente un’approssimazione in quanto, dovunque si trovi l’elettrone nella molecola, è contemporaneamente soggetto alle azioni di A e B). Questa approssimazione comporta una notevole semplificazione in quanto ci permette di considerare la situazione nei seguenti termini:
1) quando l’elettrone è più vicino al nucleo A, essendo influenzato solo da A e dagli elettroni vicini ad A, può essere descritto da una funzione d’onda ψA che è uguale alla funzione d’onda atomica che si avrebbe considerando un elettrone che si trova intorno ad un nucleo di un atomo isolato;
2) quando lo stesso elettrone è più vicino al nucleo B, essendo influenzato sole da B e dagli elettroni vicini a B, può essere descritto da una funzione d’onda ψB che e’ uguale alla funzione d’onda atomica che si avrebbe considerando un elettrone che si trova intorno ad un nucleo di un atomo isolato.
In definitiva: nella zona prossima ad A l’orbitale molecolare assomiglia all’orbitale atomico ψA ; nella zona prossima a B l’orbitale molecolare assomiglia all’orbitale atomico ψB; per l’intera molecola l’orbitale molecolare dovrà essere originato da una particolare combinazione degli orbitali atomici ψA e ψB.
Ebbene, il metodo L.C.A.O. assume che la combinazione tra ψA e yB sia una combinazione lineare, assume cioè che l’orbitale molecolare y sia originato da una combinazione lineare degli orbitali atomici ψA e ψB:
ψ = ψA + kψB
deve k è una costante che può assumere tutti i valori da – ∞ a + ∞ e che deve essere scelta, analogamente a quanto visto per a e per b nel metodo L.V., i modo da rendere minima l’energia.
Vi sono comunque delle condizioni, relative agli orbitali atomici ψA e ψB, per la formaziene dell’orbitale molecolare ψ; affinché ψA e ψB si combinino occorre che:
1) ψA e ψB abbiano energie dello stesso ordine di grandezza;
2) l’orbitale ψA si sovrapponga molto all’orbitale ψB ;
3) gli orbitali ψA e ψB abbiano la stessa simmetria rispetto all’asse della molecola (rappresentato dalla congiungente i nuclei A e B).
Il fatto che qualcuna delle condizioni precedenti non si presenti implica che gli orbitali ψA e ψB non si combinano o, al massimo, si combinano molto poco.
E’ bene precisare meglio il punto 3 riportando una tavola in cui sono elencate le combinazioni permesse e proibite da ragioni di simmetria, per alcuni orbitali che si incontrano più frequentemente: gli s, p, d (si veda la Tavola 5).
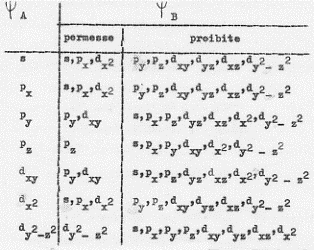
Tavola 5 (Tratta da Coulson – La valenza – Zanichelli, 1970)
Arrivati a queste punto, prima di passare alla descrizione ed allo studio della formazione di qualche molecola, esaminiamo meglio l’orbitale molecolare y incontrato qualche riga più su:
ψ = ψA + kψB
Per la costante k avevamo detto, tra l’altro, che deve essere scelta in modo da rendere minima l’energia. Ebbene, si può dimostrare che k può assumere due valori:
k = ± 1
cui corrispondono i due orbitali molecolari:
ψ+ = ψA + ψB
ψ– = ψA – ψB
Analogamente a quanto visto per il metodo L. V., l’orbitale molecolare ψ+ , corrispondendo ad un addensamento degli elettroni nella zona internucleare con conseguente schermaggio delle stesse cariche positive dei nuclei, sarà un orbitale molecolare legante a cui corrisponde un valore energetico minore(5) di quello dei singoli orbitali atomici ψA e ψB .
Al contrario, l’orbitale molecolare ψ– sarà antilegante ed avrà un valore energetico maggiore(6) di quello dei singoli orbitali atomici ψA e ψB; a questa maggiore energia corrisponderà una posizione degli elettroni non più nello spazio internucleare, con la conseguenza che non c’è più un efficace schermaggio alla repulsione tra le cariche positive dei nuclei.
Un confronto qualitativo fra le energie degli orbitali atomici ψA e ψB dell’orbitale legante ψ+ e di quello antilegante ψ– è mostrato in figura 12.
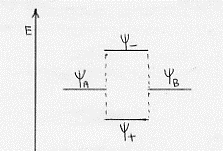
Figura 12
In definitiva, poiché ad energia più bassa corrisponde una più elevata stabilità, nella formazione di una molecola: o si ha un orbitale molecolare di tipo ψ+ (ai veda la figura 12), oppure gli elettroni che eventualmente ai trovassero nell’orbitale ψ– tenderebbero a tornare nella situazione di maggiore stabilità, tornerebbero cioè ai loro orbitali atomici yA e yB a cui compete minore energia di ψ– con conseguente non formazione della molecola. (7)
3 – Orbitali molecolari di molecole biatomiche: origine, forma e simbolismo.
Ricordiamo che nel caso atomico gli orbitali vengono indicati con s, p, d, f, … a seconda del valore del numero quantico secondario l:
| l | 0 | 1 | 2 | 3 | … |
| nome | s | p | d | f | … |
Nel caso molecolare gli orbitali vengono indicati con s, p, d, f, … (leggi: sigma, pi greco, delta, fi, …) a seconda del valore assoluto (cioè: indipendentemente dal segno) del numero quantico magnetico l (che, ricordiamolo, corrisponde al numero quantico m del caso atomico):
| l | 0 | ±1 | ±2 | ±3 | … |
| nome | s | p | d | f | … |
L’ulteriore distinzione dell’orbitale in legante o antilegante la si fa ponendo un asterisco in alto a destra del simbolo dell’orbitale antilegante. La tavola precedente può così venire integrata considerando la suddetta divisione degli orbitali in leganti o antileganti:
| l | 0 | ±1 | ±2 | ±3 | … |
| orbitali leganti | s | p | d | f | |
| orbitali antileganti | s* | p* | d* | f* | … |
D’accordo sui simboli σ, σ*, p, p*, d, d*, f, f*, …, ma quali orbitali atomici si sono combinati per originare questi orbitali molecolari ? Ad esempio, per avere un s quali orbitali atomici si devono combinare ? e per avere un p ? E’ allora necessario introdurre un simbolismo più particolareggiato che, appunto, ci dia informazioni anche sugli orbitali atomici da cui derivano gli orbitali molecolari in oggetto.
Premesso che debbono essere rispettate le 3 condizioni sugli orbitali atomici che abbiamo dato all’inizio del primo paragrafo di questa sezione, valgono le seguenti regole:
1) Se si combinano due orbitali atonici di tipo 1s si ottengono orbitali molecolari di tipo σ e σ* che, proprio per la loro origine da orbitali atomici 1s, vengono indicati con σ1s σ*1s [poiché per l = 0 si hanno orbitali molecolari di tipo σ e σ* ] in definitiva:
σ 1s ~ ψA (1s) + ψB (1s)
σ* 1s ~ ψA (1s) – ψB (1s)
2) Se si combinano due orbitali atomici di tipo 2s si ottengono orbitali molecolari di tipo σ e σ* che, proprio per la loro origine da orbitali atomici 2s, vengono indicati con σ 2s e σ*2s [poiché per l = 0 si hanno orbitali molecolari di tipo σ e σ*]. In definitiva:
σ 2s ~ ψA (2s) + ψB (2s)
σ* 2s ~ ψA (2s) – ψB (2s)
3) Se si combinano due orbitali atomici di tipo 2px si ottengono due orbitali molecolari di tipo σ e σ* . Essi vengono indicati con σ 2px e σ*2px [poiché per l = 0 si hanno orbitali molecolari di tipo σ e σ*]. In definitiva:
σ 2px ~ ψA (2px) + ψB (2px)
σ* 2px ~ ψA (2px) – ψB (2px)
4) Se si combinano due orbitali atomici di tipo 2py si ottengono orbitali molecolari di tipo π e π*. Essi vengono indicati con π2py e π*2py [poiché per l = ± 1 si hanno orbitali molecolari di tipo π e π*]. In definitiva:
π 2py ~ ψA (2py) + ψB (2py)
π* 2py ~ ψA (2py) – ψB (2py)
5) Se si combinano due orbitali atonici di tipo 2pz si ottengono orbitali molecolari di tipo π e π*. Essi vengono indicati con π2pz e π*2pz [poiché per l = ± 1 si hanno orbitali molecolari di tipo π e π*]. In definitiva:
π 2pz ~ ψA (2pz) + ψB (2pz)
π* 2pz ~ ψA (2pz) – ψB (2pz)
A questo punto si potrebbe continuare a descrivere le combinazioni dei più svariati orbitali atomici; per i fini che ci siamo proposti, però, l’esemplificazione fatta è più che sufficiente. Occorre piuttosto passare a vedere come sono fatti questi orbitali molecolari disegnandone le loro superfici limite in sezione (figura 13) ed in prospettiva spaziale (figura 14, nella quale è aggiunto un esempio di orbitale molecolare d legante – fig.14 e).
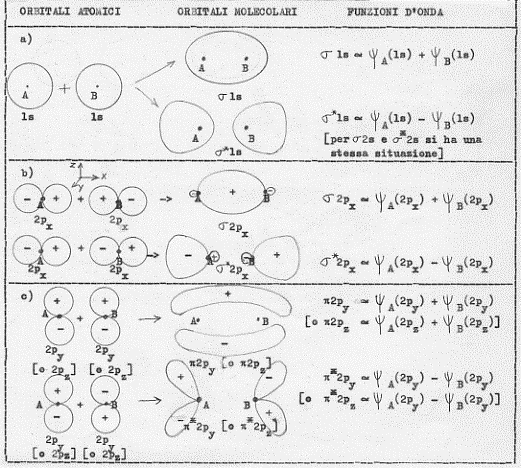
Figura 13
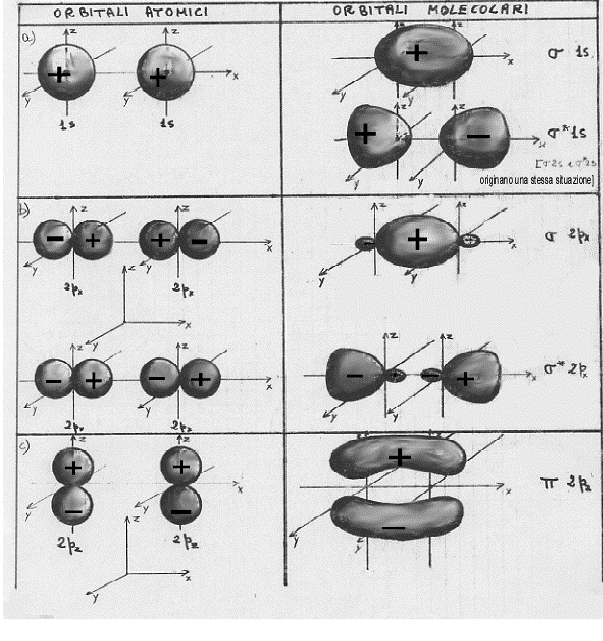
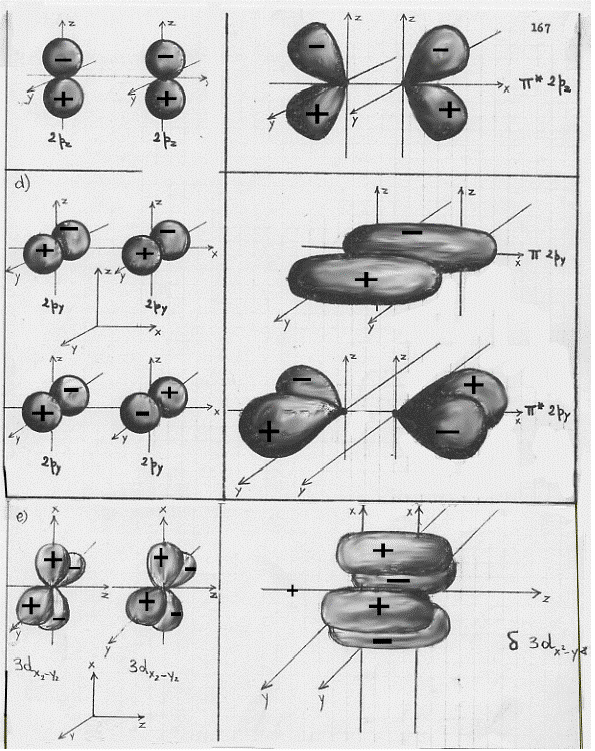
Figura 14
Facciamo ora alcune semplici considerazioni sulle figure 13 e 14:
1) confrontando la figura 13a con la 13b (la figura 14a con la 14b) si capisce subite perché la combinazione di orbitali atomici 2px origini orbitali molecolari di tipo σ e σ*, infatti gli orbitali molecolari che si ottengono nei due casi (a e b) sono praticamente uguali, hanno cioè praticamente la stessa forma;
2) confrontando la figura 13c con la 14c e la 14d si vede subito che gli orbitali molecolari π2px e π*2py sono uguali rispettivamente agli orbitali molecolari π2pz e π*2pz , differiscono infatti solo por una rotazione di 90°;
3) è necessario ribadire, sia relativamente alla figura 13 che alla figura 14, che i segni + e – che compaiono sono i segni indicanti che la funzione d’onda è positiva o negativa (non c’entrano assolutamente nulla con questioni di carica elettrica).(8)
Determinata così la forma degli orbitali molecolari, per poter applicare il principio di aufbau occorre conoscere l’ordine con cui sono sistemate le energie degli orbitali stessi. Questo lavoro fu fatto nel 1932 da Mulliken (attraverso studi di spettri molecolari) che trovò per le energie degli orbitali molecolari di molecole costituite da atomi del primo e seconde periodo della tavola periodica il seguente ordine crescente, valido con buona approssimazione:
σ 1s < σ *1s < σ 2s < σ *2s < σ 2px < π2py = π2pz < π*2py = π*2pz < σ *2px(9)
(si noti che, poiché i σ 2p ed i π2p hanno energie dello stesso ordine di grandezza, a volte invertono la loro posizione).
Un ordine analogo esiste anche per gli orbitali che vanno da σ a σ*3px ma, in questo caso, si hanno forti dubbi sulla giustezza dell’ordine stesso anche perché è difficile da questo punto (σ 3s) in poi, trovare legami puri tra orbitali atomici di tipo s, p, d, …(10)
Vediamo comunque in un grafico, con una scala per le energie puramente indicativa, la situazione energetica dei vari orbitali le cui energie sono conosciute con sufficiente sicurezza, almeno nella maggior parte dei casi (figura 15).
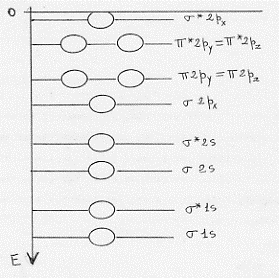
Figura 15
Prima di chiudere questo paragrafo e di passare allo studio di qualche molecola, è necessario fare ancora una piccola osservazione che il lettore più accorto avrà già fatto da sé. Perché nel caso di molecole biatomiche omonucleari non si combinano l’orbitale atomico 1s di un atomo con l’orbitale atomico 2s dell’altro ? E perché non si combinano tra loro, sempre nel caso di molecole biatomiche omonucleari, orbitali atomici di tipo s con orbitali atonici di tipo p ? La risposta è evidente e non vale nemmeno la pesa di commentarla: per atomi dello stesso elemento le energie dell’ 1s e del 2s (degli s e dei p) non sono confrontabili così come richiesto dalla prima delle tre condizioni necessarie per la formazione di orbitali molecolari.
4 – Alcune molecole biatomiche omonucleari trattate con il metodo O.M.
Abbiamo ormai tutte le carte in mano per costruirci alcune molecole biatomiche omonucleari con il metodo O.M. (facendo uso del principio di Pauli e del metodo aufbau).
H2+ – molecola-ione idrogeno.
Per formare una molecola-ione idrogeno occorre un atomo normale di idrogeno (H) ed un atomo ionizzato di idrogeno (H+ ), cioè, in pratica, un protone. L’elettrone dell’atomo normale di idrogeno ai trova nello stato 1s a più bassa energia; quando si forma la molecola questo elettrone si trasferirà nell’orbitale molecolare a più bassa energia, quello legante σ 1s. Queste fatto può essere rappresentato dalla relazione:
H (1s) + H+ = H2+[(σ 1s]
con ovvio significato dei simboli.
H2 – molecola d’idrogeno.
Ogni atomo di idrogeno che entra in combinazione per formare la molecola H2 ha l’unico elettrone nello stato 1s. Se questi due elettroni hanno spin antiparalleli, essi possono entrare, ambedue, nell’orbitale molecolare legante a più bassa energia s ls. Si ha così:
H (1s) + H (1s) = H2[(σ1s)2].
Se i due elettroni hanno spin paralleli, allora, in accordo con il principio di Pauli, un elettrone si sistemerà sull’orbitale legante s 1s e l’altro sull’orbitale immediatamente successivo, quello antilegante σ*1s:
H (1s) + H (1s) = H2 [(σ 1s)( σ*1s)].
Il fatto che ci sia un elettrone in uno stato legante ed uno in uno stato antilegante fa si che il contributo legante del σ 1s venga annullato dal contributo antilegante del σ*1s di modo che, in definitiva, se gli spin sono antiparalleli non può formarsi la molecola. Vedendo la situazione alla rovescia, se cioè per un istante dovessimo considerare una molecola H2 con un elettrone in σ 1s ed uno in σ*1s, questo fatto vorrebbe dire che la molecola normale di H2 (quella con ambedue gli elettroni in σ 1s) è stata eccitata con energia tale da permetterne la dissociazione.
Ritornando alla molecola normale (non eccitata) di idrogeno osserviamo che, poiché si hanno due elettroni nell’orbitale molecolare legante più basso, il σ 1s, questa molecola avrà un’energia di legame maggiore rispetto alla molecola-ione idrogeno H2+. Si può quiadi dire che in genere il legame a due elettroni è più forte di quello ad un elettrone.
He2+ – molecola-ione elio.
L’esistenza di questa molecola è stata rilevata da studi sugli spettri molecolari. Essa può essere formata da un atomo normale di elio He e da un atomo di elio ionizzato He+ . L’atomo normale di elio ha i suoi due elettroni nello stato ad energia più bassa 1s, così l’atomo di elio ionizzato che ha il suo unico elettrone nello stato 1s. Quando i due atomi He ed He+, entrano in combinazione per formare la molecola He2+ , i due elettroni con spin antiparalleli andranno ad occupare l’orbitale molecolare legante σ 1s, mentre il rimanente elettrone si sistemerà sull’orbitale molecolare antilegante σ*1s. Abbiamo così la seguente relazione:
He (1s2) +He+(1s) = He2+[(σ 1s)2 (σ*1s)].
La situazione che qui si presenta è quasi analoga a quella della molecola H2 . C’è in più un elettrone nell’orbitale antilegante σ*1s. Poiché da una serie di misure sperimentali si è dimostrato che l’effetto di un elettrone su di un orbitale antilegante è maggiore di quello di un elettrone su di un orbitale legante, la stabilità della molecola He2+ è minore di quella della molecola H2+. Questo spiega perché in natura si osservano molte molecole H2+ e, invece, per osservare molecole He2+ ci si debba servire di metodi spettroscopici.
He2 – molecola di elio.
La molecola di elio non esiste in condizioni normali; essa si forma solo in particolari condizioni, quando l’elio è eccitato (è stata infatti rivelata con metodi spettroscopici all’interno dei tubi di scarica) ed è, in ogni caso, instabile. La sua formazione dovrebbe essere descritta nel modo seguente:
He (1s2) + He (1s2) = He2 [(σ 1s)2(σ*1s)2]
e, come si vede, i quattro elettroni dei due elii atomici vanno a sistemarsi, nella molecola, due sull’orbitale molecolare legante σ 1s e due sull’orbitale molecolare antilegante σ*1s. In termini energetici ciò significa (si riveda la figura 15) che, poiché l’energia repulsiva dell’orbitale antilegante è maggiore di quella attrattiva dell’orbitale legante, in pratica non si ha nessun legame, quest’ultimo essendo piuttosto sostituito da una repulsione tra i singoli atomi.
Quando invece gli atomi di elio sono eccitati (quando cioè ciascun atomo ha un elettrone nell’orbitale atomico 1s e l’altro nel 2s) allora si ha la seguente situazione:(11)
He[(1s)(2s)] + He[(1s)(2s)] = He2[(σ 1s)2(σ 2s)2]
e, cose si vede, i quattro elettroni si trovano su orbitali molecolari leganti, di modo che la molecola può esistere in quello stato.
E’ utile osservare che una delle affermazioni che abbiamo ora fatto ha validità generale: quando si ha a che fare con orbitali leganti ed antileganti riempiti ciascuno dei due elettroni loro spettanti, non si può formare alcun legame.
Li2 – molecola di litio.
Il litio atomico ha tre elettroni: due nell’orbitale 1s ed uno nel 2s. L’unico elettrone di valenza è quello che si trova nell’orbitale 2s. La molecola di litio si formerà quindi nel modo seguente:
Li[(1s2)(2s)] + Li[(1s2)(2s)] = Li2[KK(σ 2s)2]
dove, l’aver introdotto il K in luogo degli ipotetici (σ1s)2 e (σ*1s)2, vuol dire che il livello 1s del del singolo atomo non entra a far parte del legame molecolare e pertanto non si formano orbitali molecolari con i singoli 1s. Questi orbitali atomici, essendo completi, rimangono tali anche all’interno della molecola e quindi risultano indicati con il nome dello strato atomico cui si riferiscono che è, appunto, il K.
Il legame si forma allora solo con l’orbitale molecolare σ 2s e, poiché gli orbitali atomici 2s distano dal nucleo circa 4 volte di più degli orbitali 1s, la distanza esistente tra i due nuclei di litio nella molecola sarà sensibilmente maggiore della distanza tra i due nuclei di idrogeno nella molecola H2 . Ciò comporta una minore sovrapposizione e conseguentemente una minore energia di dissociazione per la molecola (circa 1/4 di quella necessaria per dissociare H2).
B2 – molecola di boro.
Il boro atomico ha 5 elettroni: due nell’orbitale 1s, due nell’orbitale 2s ed uno nel 2p. La molecola B2 ha quindi 10 elettroni che, ricordando Pauli e le regole di Hund, ai sistemeranno nel modo seguente negli orbitali molecolari:
B[(1s2)(2s2)(2p)] + B[(1s2)(2s)2(2p)] = B2[KK(σ 2s)2(σ*2s)2(π 2py)(π 2pz)].
Poiché nel caso della molecola in esame, come in altri casi (C2 , N2+ , N2 ) ed in genere per molecole biatomiche omonucleari formate da elementi del 2° periodo, le energie dei vari orbitali molecolari hanno valori molto vicini, la struttura dei successivi riempimenti elettronici è un poco incerta e comunque non è quella che si dovrebbe avere semplicemente prendendo in esame la successione energetica dei vari orbitali mostrata in figura 15 (che, ricordiamolo, non è certa per tutte le molecole ma solo quella più volte realizzata). Non si ha quindi, come ci si sarebbe aspettati, un orbitale (σ 2px)2 pieno, ma, al contrario, si hanno due elettroni disaccoppiati sui due orbitali p leganti ad uguale energia (degeneri). E ciò è provato da misure con metodi spettroscopici.
Poiché, come nel caso del litio, gli orbitali atomici 1s non entrano a far parte del legame molecolare, si ha a che fare con 4 elettroni leganti e due antileganti; c’è quindi un effetto risultante di 2 elettroni leganti corrispondente ad un legame singolo.
Nella Tavola 6 sono riportate le configurazioni elettroniche di svariate molecole biatomiche omonucleari (negli stati fondamentali, cioè non eccitati), alcune delle quali le abbiamo già incontrate ed altre le incontreremo nelle pagine seguenti.
N2 – molecola di azoto.
L’atomo di azoto ha 7 elettroni e, in accordo con le regole di Hund ed il principio di Pauli, presenta la seguente configurazione elettronica:
N[(1s)2(2s)2(2px)(2py)(2pz)].
La molecola N2 avrà quindi 14 elettroni. Anche qui, come nei caso del litio e del boro, gli orbitali atomici 1s non entrano a far parte del legane e, pertanto, la molecola sarà costruita nel modo seguente:
2N[(1s2)(2s2)(2px)(2py)(2pz)] = N2[KK(σ 2s)2(σ *2s)2( π2py = π2pz)4(σ 2px)2].
L’effetto legante del (σ 2s)2 sarà praticamente annullato dall’effetto antilegante del (σ*2s)2 , di modo che si avranno solo 6 elettroni in stati leganti. Conseguentemente si ha a che fare con un triplo legame: uno di tipo s e due di tipo p. Questo legame è il più forte possibile per una molecola biatomica omonucleare e perciò N2 risulta molto stabile (si veda la Tavola 6. Come esemplificazione, nella figura 16, seno illustrati gli orbitali molecolari della molecola N2 .

.TAVOLA 6 (Rielaborazione da Herzberg: Molecular Spectra … – Prentice-Hall, 1939).
Si noti che per ordine del legame si intende la quantità così definita: numero degli elettroni la orbitali molecolari leganti meno il numero degli elettroni in orbitali molecolari antileganti; quello che risulta va diviso per due. [I valori numerici segnati con asterisco sono incerti].
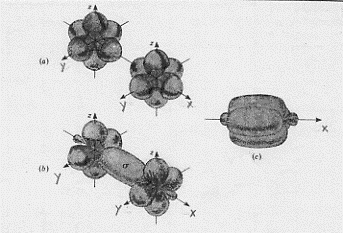
Figura 16 – Orbitali molecolari della molecola di azoto.
In (a) sono mostrati i tre orbitali p per i due atomi , in (b) è mostrata la formazione dell’orbitale molecolare s lungo l’asse x; in (c) sono mostrati gli orbitali molecolari di tipo p che si formano per sovrapposizione degli orbitali atomici lungo gli assi y e z.
O2 – molecola di ossigeno.
L’atomo di ossigeno ha 8 elettroni nella seguente configurazione:
O[(ls2)(2s2)(2px)(2py)(2pz)2]
La molecola O2 dispone allora di due elettroni in più rispetto a quelli di cui disponeva l’azoto e questi elettroni si andranno a disporre nell’orbitale molecolare antilegante p* con modalità che discuteremo tra un poco. La configurazione elettronica di O2 sarà allora:
2O[(1s2)(2s2)(2px)(2py)(2pz)2] = O2[KK(σ 2s)2(σ *2s)2(σ 2px)2(π2py = π 2pz)4(π*2py = π*2pz)2]
Ora, poiché gli orbitali π sono degeneri (il π 2py ha la stessa energia del π 2pz) analogamente ai π* , mentre per i π disponiamo di 4 elettroni che li vanno a saturare, per i π* si hanno solo 2 elettroni che, in accordo con le regole di Hund, andranno a disporsi con gli spin paralleli, uno in π*2py ed uno in π*2pz .
La molecola O2 ha quindi 2 elettroni disaccoppiati (con spin paralleli). Questi elettroni disaccoppiati rendono conto del fatto sperimentale che la molecola di ossigeno ha particolari proprietà magnetiche (è paramagnetica). E la spiegazione così elegante di questo fenomeno (insieme ad una analoga spiegazione del paramagnetismo della molecola S2 di zolfo)(12), fornita dalla teoria O.M., costituì uno dei suoi primi successi (la teoria del legame di valenza non prevedeva l’esistenza di due elettroni disaccoppiati e quindi non spiegava il paramagnetismo dell’ossigeno).
Nella figura 17 è riportata in modo schematico la formazione della molecola O2 , a partire dai due atomi O.
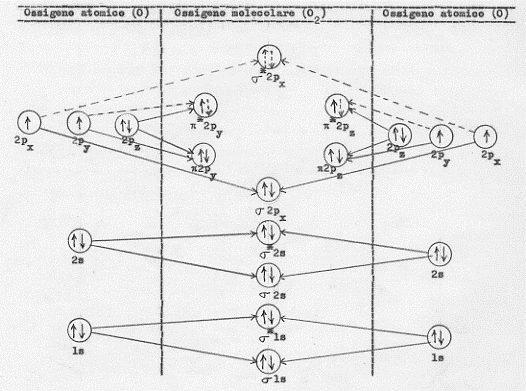
Figura 17. Le transizioni a tratto continuo sono quelle che realmente hanno luogo quelle tratteggiate sono quelle che si avrebbero nel caso i 2px ed i 2py avessero due elettroni.
Riguardo al legame, ora, rispetto alla molecola N2, abbiamo due elettroni la più in uno stato antilegante che andranno praticamente ad annullare il contributo di due elettroni in uno stato legante; si avranno così 4 elettroni in uno state legante con la conseguenza che si ha un doppio legame.
*****
Per le molecole F2 ed Ne2 , non essendoci novità di interesse, rimando alla Tavola 6.
*****
5 – Altre molecole biatomiche omonucleari.
Quanto abbiamo visto per le molecole biatomiche omonucleari del secondo periodo della Tavola periodica può essere esteso alle molecole biatomiche omonucleari qualunque. Naturalmente occorrerà stabilire con precisione il valore del numero quantico n degli orbitali di valenza e bisognerà tener conto di una maggiore incertezza nell’ordine delle energie crescenti degli orbitali molecolari.
In modo del tutto generale si può dire che le molecole biatomiche omonucleari dei metalli alcalini, come Na2 , K2, Rb2 , Cs2 (Gruppo 1A della Tavola Periodica degli elementi riportata in 39 – 7), hanno configurazioni del tipo visto per la molecola Li2 . Il legame avviene con un orbitale molecolare di tipo s e non si hanno elettroni disaccoppiati. Ad esempio, la molecola di sodio (Na2) ha la seguente configurazione:
Na2[KK,LL,(σ 3s)2]
là dove LL ha lo stesso significato di KK e cioè che gli elettroni atomici degli strati L dell’atomo di sodio non prendono parte al legame. Tutte le molecole di questo tipo esistono in fase di vapore ed hanno proprietà diamagnetiche.
Allo stesso modo le molecole biatomiche omonucleari formate da atomi del 5° gruppo (A) della Tavola periodica; esse hanno una configurazione del tipo visto per la molecola N2 dell’azoto. Il legame è di tipo s e p e non vi sono elettroni disaccoppiati. Ad esempio, la molecola P2 di fosforo ha gli strati K ed L pieni, mentre il legame si stabilisce con gli orbitali molecolari (σ 3p)2 e (π3py = π3pz)4 .
Infine, le molecole biatomiche omonucleari degli alogeni, come Cl2 ,Br2 , I2 (Gruppo 7A della Tavola periodica), hanno configurazioni elettroniche del tipo di quella della molecola F2 di fluoro (si veda la Tavola 6). Il legame è di tipo s e le molecole risultano diamagnetiche.
NOTE
(1) Ricordiamo brevemente le limitazioni dei numeri quantici e spieghiamo perché m è sostituito, per le molecole, da l. Si ha:
n = 1, 2, 3, … , ∞
l = 0, 1, 2, … , n – 1
λ = 0, ±1, ±2, … , ± l
e, come si vede le cose stanno proprio come nel caso atomico. La sostituzione di m con λ si spiega subito ricordando che, nel caso atomico, m era il numero quantico che ci forniva la quantizzazione spaziale (il fatto cioè che l’orbita elettronica poteva essere orientata, rispetto ad un arbitrario a sse di riferimento, solo lungo certi assi stabiliti, appunto, da m); si capisce bene che, nel caso molecolare (di una molecola biatomica, per fissare le idee), l’asse di riferimento non è più arbitrario ma sarà la linea congiungente i due nuclei: questo semplice fatto comporta un cambiamento di notazione da m a λ .
(2) L’abbreviazione L.C.A.O. sta per Linear Combinations of Atomic Orbitals ed è dovuta a Mulliken (1935).
(3) Ricordo che più è basso il valore dell’energia più il legame di una molecola è stabile.
(4) Le funzioni d’onda ψ+ e ψ– , nella teoria dell’orbitale molecolare, sono molto spesso indicate con ψg e ψu . I subindici g ed u sono le iniziali delle parole tedesche gerade (= simmetrica) e ungerade (= antisimmetrica) con riferimento appunto a proprietà di simmetria o antisimmetria spaziale delle stesse funzioni d’onda rispetto al centro della molecola (si veda più oltre). Allo scopo ricordiamo che una funzione f (x, y, z) è spazialmente simmetrica se risulta:
f (x, y, z) = f (-x, -y, -z);
è invece spazialmente antisimmetrica se risulta:
f ( x, y, z) = – f (-x, -y, -z).
Se x, y, z sono le coordinate di un elettrone è evidente l’estensione ad orbitali molecolari simmetrici o antisimmetrici di quanto detto per una generica funzione f(x, y, z).
(5) E proprio per questo l’orbitale molecolare è legante e, se sono rispettat e le condizioni: di energie di yA e yB confrontabili, di massima sovrapposizione di yA e yB e di simmetria di yA e yB rispetto all’asse molecolare originerà una molecola stabile.
(6) In definitiva, tra i due orbitali molecolari y+ e y– quello cui compet e energia minore è y+ e di conseguenza è l’unico orbitale molecolare da prendere in considerazione per la formazione di una molecola stabile (sempre se sono rispettate le condizioni ricordate nella nota precedente).
(7) Si possono trovare elettroni (al massimo 2, in accordo con Pauli) su un orbitale antilegante di tipo y– solo quando l’orbitale legante è già pieno con i due elettroni che gli competono.
(8) Facciamo ora una considerazione complementare alla nota 4 nella quale abbiamo parlato di funzioni d’onda ψg simmetriche e yu antisimmetriche rispetto al centro della molecola.
Premesso che simmetria e antisimmetria di un orbitale molecolare non devono essere confuse con il suo essere legante o antilegante (ci sono orbitali leganti sia simmetrici che antisimmetrici e, viceversa, ci sono orbitali antileganti sia simmetrici che antisimmetrici) studiamo un poco meglio la questione.
Cominciamo con il definire come asse molecolare la retta congiungente i due nuclei di una molecola biatomica:
A————B
Definiamo poi come centro della molecola il punto C che si trova a metà di A e B. Considerando ora un generico orbitale molecolare, si prenda una qualsiasi retta passante per il centro C della molecola. Questa retta incontrerà l’orbitale molecolare in due punti giacenti su semipiani opposti rispetto alla retta passante por A e B. Se in tali punti (chiamiamoli 1 e 2) la funzione d’onda ψ che descrive l’orbitale molecolare ha lo stesso segno, allora l’orbitale sarà simmetrico (g); se il segno è opposto allora l’orbitale sarà antisimmetrico (u). Operando in questo modo andiamo a studiarci la simmetria o antisimmetria degli orbitali molecolari che conosciamo (si riveda la figura 13):
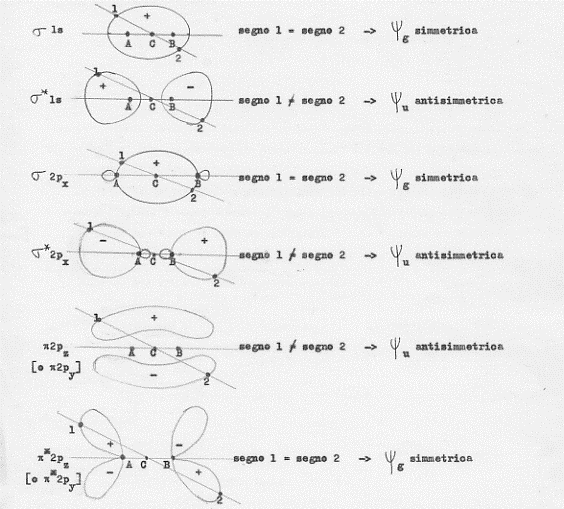
Un’ultima nota sulla transizione degli elettroni da uno stato ad un altro. Si può dimostrare che sono possibili solo transizioni da uno stato g ad uno stato u e, viceversa, da uno stato u ad uno stato g; non sono invece possibili transizioni tra stati g e tra stati u:
| transizioni permesse | g ->u | u ->g |
| transizioni proibite | g ->g | u ->u |
(9) Lo stesso Mulliken suggerì (1932) un’altra notazione, equivalente a quella data:
(K)zσ < (K)yσ < zσ < yσ < xσ < wπ = wπ < vπ = vπ < uσ
da cui si ricavano le seguenti identità:
(K)zσ = σ
(K)yσ = σ*
zσ = σ 2s
yσ = σ*2s
xσ = σ 2px
wπ = π 2py = π 2pz
vπ = π*2py = π*2pz
uσ = σ*2px.
Sussistono poi anche queste altre identità:
(m)z = σ 3s
(m)y = σ*3s
(m)x = σ 3px
(m)wπ = π 3py = π 3pz
(m)vπ = π*3py = π 3pz
(m)u = σ*3px
(10) In ogni caso, la sequenza più probabile sembra essere:
σ 3s < σ*3s < π 3py = π 3pz < σ 3px < π*3py < π*3pz < σ*3px .
(11) Si noti che alcune regole dette di selezione (che non abbiamo studiato) impediscono un passaggio diretto dell’elettrone dall’orbitale 1s al 2s (deve risultare Dl = ± 1 ). E’ però possibile, ad esempio, che l’elettrone sia eccitato passando dal livello 1s al 3p e quindi, diseccitandosi parzialmente, passi dal 3p al 2s. Inoltre, una volta che l’elettrone è arrivato al 2s con il meccanismo ora esemplificato, poiché ancora le regole di selezione impediscono che dal 2s esso possa passare al livello 1s, lo stato 2s rimane occupato più del dovuto (stato metastabile) ed è quello, appunto, che permette il legame molecolare. Infine, all’interno di un campo elettrico sono possibili transizioni tra il livello 1s ed il 2s (e viceversa) anche se, in questo caso, risulta Dl = 0.
(12) Si noti che a temperature fino a circa 800 °C si ha a che fare con molecole di zolfo S8; tra gli 800 °C ed i 2.000 °C si ha a che fare con molecole S2 di zolfo; al di sopra dei 2.000 °C si hanno atomi di zolfo.
1 – Molecole biatomiche eteronucleari trattate con il metodo O.M.
Quando si ha a che fare con molecole biatomiche eteronucleari, molecole formate da due atomi di due elementi diversi (come, ad esempio, acido fluoridrico HF, acido cloridrico HCl, monossido di carbonio CO, ossido di azoto NO, idruro di litio LiH, … ), si può ancora applicare il metodo O.M. nell’approssimazione L.C.A.O. con alcune avvertenze. Innanzitutto, quando si fa la combinazione lineare degli orbitali atomici (L.C.A.O.) tra l’orbitale y A dell’atomo A e l’orbitale yB dell’atomo B per ottenere l’orbitale molecolare y :
ψ = ψA + k ψB
il k non assume più i valori ± 1, che abbiamo visto assumere nel caso di molecole biatomiche omonucleari. Questa volta occorrerà farsi i conti di volta in volta, a seconda di quali sono gli orbitali atomici in giuoco. Infatti, mentre nel caso omonucleare gli orbitali atomici ψ A e ψB erano sempre dello stesso tipo (o 1s, o 2s, o 2px , …), ora ψ A sarà generalmente differente da ψB (ad esempio: il primo 1s ed il secondo 2p).(1) Si avranno comunque due valori di k, uno del quali relativo ad un orbitale molecolare legante e l’altro ad un orbitale molecolare antilegante. Per rendere conto di ciò è conveniente a volte indicare la relazione precedente nel modo seguente:
ψ = k1ψA + k2ψB
là dove il rapporto k2/k1 fornisce il k precedentemente introdotto.
Valgono invece le altre condizioni e cioè che:
1) ψA e ψB debbono avere energie dello stesso ordine di grandezza;
2) l’orbitale ψA abbia la massima sovrapposizione con yB;
3) gli orbitali ψA e ψB abbiano una stessa simmetria rispetto all’asse della molecola.
Con queste condizioni è facile stabilire quali sono gli orbitali atomici ψ A e ψB che vanno a formare l’orbitale molecolare ψ. (2)
Vale ancora la classificazione in orbitali molecolari leganti ed antileganti ed in generale risulta (Mulliken, 1932):(3)
(k)z σ <(k)yσ < zσ < wπ = wπ < yσ < xσ < vπ = vπ < uσ ;
non si dovrà invece più considerare la classificazione delle funzioni d’onda in simmetriche ed antisimmetriche poiché ora la molecola, formata da orbitali atomici differenti, non avrà più un centro di simmetria. C’è infine da notare che, a seguito della diversità della carica posseduta dai due nuclei dei due atomi che vanno a formare la molecola, i livelli energetici (o orbitali) dello stesso tipo avranno energie differenti nei due atomi (ad esempio: il livello 1s dell’idrogeno ha energia differente dal livello 1s del fluoro).
Possiamo ora passare a descrivere la formazione di qualcuna di queste molecole.
HF – Molecola di acido fluoridrico.
Gli atomi di idrogeno e fluoro hanno rispettivamente le seguenti configurazioni:
H [(1s)] ; F [(1s2)(2s2)(2px)(2py2)(2pz2)].
Gli elettroni che si trovano negli orbitali 1s e 2s del fluoro non entrano a far parte del legame e restano nella situazione di orbitali atomici del fluoro. Negli orbitali 2p vi sono invece 5 elettroni e quindi uno di essi risulterà disaccoppiato (nella configurazione del fluoro che abbiamo dato è l’orbitale 2px che risulta privo di un elettrone; ma era indifferente considerare che l’elettrone mancasse al 2py o al 2zp). L’aver scelto l’orbitale 2px privo di un elettrone significa che è proprio questo orbitale che va a combinarsi in orbitale molecolare con un orbitale atomico 1s dell’idrogeno. E proprio perché abbiamo scelto il 2px sarà* necessario che l’asse della molecola (che unisce il nucleo dell’atomo di idrogeno con quello di fluoro) si trovi lungo l’asse x in modo da rispettare, per una data distanza tra i nuclei, la regola della massima sovrapposizione (figura 18).(4) La sovrapposizione tra gli orbitali atomici 1s dell’idrogeno e 2px del fluoro (il discorso per il 2pz è esattamente identico) avrebbe originato la situazione di figura 19. Come si può vedere da un confronto con la figura 18a, risulta una sovrapposizione (zona tratteggiata) minore ed inoltre, mentre nel caso della figura 18 si ha sovrapposizione di parti di orbitale con lo stesso segno, ora si ha a che fare con una sovrapposizione con parti di orbitale che hanno segno opposto e questa sovrapposizione risulta perfettamente equilibrata di modo che i due
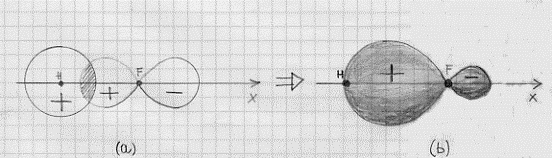
Figura 18
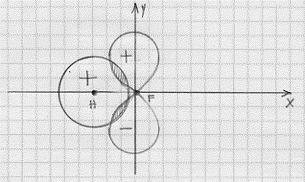
Figura 19
contributi si annullano vicendevolmente.
In definitiva anche gli orbitali 2py e 2pz rimangono come orbitali atomici e la configurazione della molecola sarà:
H[(1s)] + F[(ls2)(2s2)(2px)(2py2)(2pz2)] = HF[K(2s2)(xσ)2(2py2)(2pz2)].
L’orbitale molecolare cui si deve ti legame è quindi di tipo σ (5) e, per quanto detto in apertura del paragrafo, sarà dato da:
ψ(s 2px) = k1(1s) + k2 ψ(2px).
Poiché poi, elaborando le misure sperimentali,(6) si trova che k2 è maggiore di k1 e, che è lo stesso, k = k2 /k1 è maggiore di uno, ciò vorrà dire che il contributo relativo all’orbitale molecolare dell’orbitale atomico 2px è superiore al contributo relativo fornito dall’orbi tale atomico 1s. La ragione di ciò risiede nel fatto che i due orbitali atomici che si uniscono per formare l’orbitale molecolare hanno energie diverse risultando, in questo caso, superiore l’energia del 2px del fluoro di quella dell’orbitale 1s dell’idrogeno. Ed in definitiva, a seguito del fatto che k2 > k1 , i due elettroni che prendono parte al legame tenderanno a trovarsi per la maggior parte del loro tempo più vicini al nucleo del fluoro (la densità di carica della nube elettronica sarà maggiore vicino al fluoro che non all’idrogeno) che fornisce un orbitale di valenza più stabile di quello fornito dall’idrogeno. Ciò si può anche dire affermando che l’atomo di fluoro è più elettronegativo(7) di quello dell’idrogeno. I due coefficienti k1 e k2 , legati da k = k2 /k1 , sono quindi molto importanti poiché, in definitiva, si forniscono il grado di polarità di una molecola (la molecola avrà un carattere ionico parziale).
*****
Con m meccanismo del tutto simile a quello ora visto per l’acido fluoridrico, formano molecole tutti i composti dell’idrogeno con gli alogeni (cloro, bromo, iodio, … ). Vediamo rapidamente l’esempio dell’acido cloridrico (HCl).
HCl – molecola di acido cloridrico.
Gli atomi di idrogeno e cloro hanno le seguenti configurazioni:
H[(1s)]; Cl [(1s2)(2s2)(2px2)(2py2)(2pz2)(3s2)(3px)(3py2)(3pz2)].
Gli elettroni del cloro che si trovano sugli orbitali 1s (strato K), 2s, 2px , 2py, 2pz (strato L) non entrano a far parte del legame e restano nella situazione di orbitali atomici del cloro. L’orbitale 3px del cloro è privo di un elettrone e quindi sarà questo l’orbitale atomico che andrà a formare, con l’orbitale atomico 1s dell’idrogeno, l’orbitale molecolare legante di tipo s. La configurazione elettronica della molecola HCl sarà quindi:
H[(1s)] + Cl [(1s2)(2s2)(2px2)(2py2)(2pz2)(3s2)(3px)(3py2)(3pz2)] = HCl [KL(3s2)(mxs)2(3py2)(3pz2)]
Anche qui, poiché risulta k2 > k1 si avrà che il contributo relativo dell’orbitale atomico 3px all’orbitale molecolare mx s è superiore al contributo relativo fornito dall’orbitale atomico 1s. E poiché k2 > k1 , i due elettroni che prendono parte al legame tenderanno a stare più vicini all’atomo di cloro che non all’atomo di idrogeno. Ciò vuol dire che l’atomo di cloro è più elettronegativo di quello di idrogeno e che la molecola ha caratteristiche polari.
LiH – molecola di idruro di litio.
Il litio ha tre elettroni:
Li[(1s2)(2s)]
ma, poiché l’orbitale 2s ha un’energia che è molto vicina a quelle degli orbitali 2p, sono orbitali di valenza sia il 2s che i 2p.(8) Anche qui però si può procedere con buona approssimazione considerando che la molecola LiH nasca dalla sovrapposizione dell’orbitale 1s dell’idrogeno con il 2s del litio (il livello 1s del litio risulta infatti più stabile del livelli 2p) come mostrato in figura 20. Il legame risulta quindi di tipo s e la configurazione elettronica di LiH sarà:
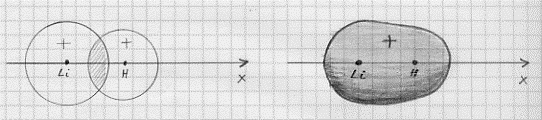
Figura 20
H[(ls)] + Li[(1s2)(2s)] = LiH[K(zs)2].
Poiché poi, in questo caso, risulta k1 > k2 e cioè k < 1, i due elettroni che formano il legame verranno a trovarsi più vicini all’atomo di idrogeno che non all’atomo di litio (carattere ionico parziale della molecola che ha caratteristiche polari). L’idrogeno risulta quindi più elettronegativo del litio.
CO — molecola di monossido di carbonio.
La struttura di questa molecola ha fatto molto discutere, a partire da Mulliken (1932) fino a Sahni (1953).
Le configurazioni elettroniche degli atomi di carbonio e di ossigeno sono rispettivamente:
C[(1s2)(2s2)(2px)(2py)] ; O[(1s2)(2s2)(2px)(2py)(2pz2)]
Come si vede, sia nell’uno che nell’altro atomo vi sono due orbitali da completare e precisamente i 2px ed i 2py ; inoltre al 2pz dell’ossigeno, che è completo, corrisponde un 2pz del carbonio che è vuoto. In accordo con l’elaborazione di Mulliken, la molecola CO avrà la seguente configurazione:
C[(1s2)(2s2)(2px)(2py)] + O[(1s2)(2s2)(2px)(2py)(2pz2)] = CO[KK(zs)2(ys)2(xs)2(wp)4] (9)
Gli orbitali leganti sono zs, xs e wp; sono cioè in numero di 4, mentre si ha un solo orbitale antilegante, lo ys. In definitiva si ha a che fare con un triplo legame; uno di tipo s e due di tipo p. La situazione secondo Mulliken è analoga a quella che si presenta nella molecola N2 di azoto.(10) Ma proprio da questa analogia sorge una difficoltà: infatti, ionizzando una molecola N2 di azoto per ottenere N2+ , si ha una diminuzione dell’energia di legame, contrariamente a quello che accade ionizzando CO per ottenere CO+ (in questo caso l’energia di legame aumenta).(11)
Questo fatto può essere compreso solo ammettendo che il legame, doppio nella molecola CO non ionizzata, diventa triplo nella molecola CO + ionizzata (contrariamente a quanto avviene per l’azoto).
Come accordare le due conclusioni ? Come accordare, cioè, il fatto che da una parte risulta un legame triplo e dall’altra un legame doppio ?
Secondo Long e Walsh (1947) è possibile pensare che quando C ed O si avvicinano per formare la molecola, mentre gli orbitali atomici 1s di ambedue rimangono orbitali atomici e mentre gli orbitali atomici 2s di ambedue origineranno gli orbitali molecolari zs ed ys (rispettivamente legante ed antilegante), l’orbitale 2pz dell’ossigeno, che risulta pieno, resterà prevalentemente come orbitale atomico dell’ossigeno, interagendo però debolmente con l’orbitale 2pz del carbonio che risulta vuoto. Ciò vuol dire che uno degli orbitali molecolari wp avrà essenzialmente un carattere atomico, risultando localizzato principalmente intorno all’ossigeno. Questo tipo di legame, composto da due legami netti e da uno fornito essenzialmente dall’atomo di ossigeno, lo si usa indicare nel modo seguente (doppio legame covalente + un debole legame d’altra natura):
in ogni caso, ancora oggi non abbiamo elementi sufficienti per affermare che quanto qui sostenuto sia esente da dubbi.
A questo punto resta solo da dire che la molecola CO presenta una polarità praticamente trascurabile (e questo pare in accordo con quanto sostenuto da Long e Walsh).
NO – molecola di ossido di azoto.
Molto in breve descriviamo la formazione di NO:
K[(1s2)(2s2)(2px)(2py)(2pz)] + O[(1s2)(2s2)(2px)(2py)(2pz2)] = NO[KK(zs)2(ys)2(xs)2(wp)4(vp)].
Poiché zs, xs e wp (considerato due volte) sono leganti ed ys con vp sono antileganti, l’ordine del legame sarà 2 e 1/2 (si riveda Tavola 6). La molecola è molto stabile (anche se meno di CO e di N2) e la cosa potrebbe sembrare strana per una molecola che ha un elettrone disaccoppiato. In realtà il fatto può essere spiegato semplicemente osservando che l’elettrone singolo non si trova su un orbitale atomico ma su un orbitale molecolare la cui densità di carica è uniformemente distribuita tra i due atomi, piuttosto che essere localizzata più vicino all’uno che all’altro.(12)
Da ultimo c’è da dire che l’elettrone disaccoppiato è responsabile delle proprietà paramagnetiche che presenta la molecola NO.
2 – Molecole poliatomiche trattate con il metodo L.V.: l’ibridizzazione.
L’atomo di carbonio ha la seguente configurazione:
C[(1s2)(2s2)(2px)(2py)].
Avendo due orbitali incompleti la valenza del carbonio è 2. Molto spesso però il carbonio risulta tetravalente (come nella molecola CH4 di metano). Come è possibile rendere conto di ciò ? Si può pensare di eccitare l’atomo di carbonio in modo che un elettrone 2s passi nell’orbitale 2pz (originariagieate vuoto). In questo modo il carbonio eccitato viene ad avere la configurazione seguente:
C[(1s2)(2s)(2px)(2py)(2pz)].
e, come si vede, poiché sono ora 4 gli orbitali incompleti, l’atomo è diventato tetravalente; ciascuno dei 4 orbitali può formare un legame: tre di questi legami si formeranno con orbitali di tipo p ed uno con orbitali di tipo s. Per quanto sappiamo, risulterebbe che tre dei legami avrebbero caratteristiche differenti dal quarto(13) ed invece, sperimentalmente, tutti e 4 i legami risultano con le stesse caratteristiche.
Allo stesso modo per l’atomo di boro:
B[(1s2)(2s2)(2px)]
esso sembrerebbe monovalente dato che ha il solo orbitale 2px incompleto. In realtà il boro presenta a volte caratteristiche trivalenti, come nella formazione della molecola di trifluoruro di boro (BF3). Per rendere conto di ciò si può operare come visto per il carbonio, eccitando l’atomo di boro:
B[(1s2)(2s)(2px)(2py)].
ed a tre orbitali incompleti corrisponde un atomo trivalente. Si dovrebbero allora avere 2 legami con orbitali di tipo p ed un legame con orbitali di tipo s, i primi due con caratteristiche differenti dal terzo. Ma, sperimentalmente, si è mostrato che i tre legami hanno tutti le stesse caratteristiche.
Altro esempio è il berillio:
Be[(1s2)(2s2)];
sembrerebbe che esso non debba legarsi in molecole stabili per comportarsi invece come l’atomo di elio (visto che non ha elettroni disaccoppiati).(14) Invece il berillio presenta un comportamento bivalente formando molecole stabili (come BeO, BeCl2, BeH). Anche qui si può pensare di eccitare l’atomo di berillio per ottenere:
Be[(1s2)(2s)(2px)]
In questo modo l’atomo è diventato bivalente e può formare due legami: uno con l’orbitale di tipo s ed uno con l’orbitale di tipo p. Si dovrebbe quindi avere un legame con caratteristiche differenti dall’altro e, anche qui sperimentalmente, si mostra invece che le caratteristiche dei due legami sono identiche.
Di esempi come quelli descritti ve ne sono moltissimi e per rendere conto di essi Pauling (1931) introdusse il concetto di ibridizzazione, nell’ambito dello sviluppo del metodo L.V.
All’interno del medesimo atomo, alcune volte, occorre non fare più riferimento ad orbitali s distinti da orbitali p (ed in genere bisogna abbandonare qualsiasi distinzione tra orbitali atomici). Occorre invece prendere in considerazione una sorta di mescolamento (o ibridizzazione) tra questi orbitali tale da originare orbitali atomici differenti, appunto, dagli s e p che conosciamo.
Per quel che riguarda il carbonio e per rendere conto della sua tetravalenza, si deve assumere che avvenga un mescolamento tra un orbitale s e tre orbitali p: da queste mescolamento si originano quattro nuovi orbitali atomici che, con ovvio significato dei simboli, sono chiamati ibridi sp3 (tutti perfettamente equivalenti).
Per quel che riguarda la trivalenza del boro, si deve ammettere un mescolamento tra un orbitale s e due orbitali p: da questo mescolamento si originano tre nuovi orbitali atomici che sono chiamati ibridi sp2 (tutti perfettamente equivalenti tra di loro).
Per quel che riguarda la bivalenza del berillio, si deve ammettere un mescolamento tra un orbitale s ed un orbitale p: da questo mescolamento si originano due nuovi orbitali atomici che sono chiamati ibridi sp (tutti perfettamente equivalenti tra di loro).
Alcune osservazioni vanno subito fatte: vi sono altri tipi di ibridizzazione oltre a quelli qui esemplificati (si veda la Tavola 7); le ibridizzazioni esemplificate non riguardano sole gli atomi specifici che abbiamo preso in considerazione

Tavola 7
(carbonio per l’sp3 , boro per l’sp2 , berillio per l’sp) ma hanno una validità generale; occorre sottolineare che l’ibridizzazione è un fatto atomico che si costruisce prima che si vada a formare la molecola; è molto importaste osservare che l’ibridizzazione degli orbitali atomici non implica un cambiamento dello stato degli elettroni negli atomi ma, piuttosto, una differente forma matematica nella quale gli orbitali atomici vengono descritti; non è più necessario far riferimento agli assi x, y, z.
Nella figura 21 sono mostrate le forme che assumono le superfici limite di alcuni orbitali ibridi (detti anche orbitali equivalenti): prima l’sp, quindi l’sp2 ed infine l’sp3.
In definitiva si può dire:
1) l’ibridizzazione consiste nel mescolamento di differenti orbitali atomici ;
2) si possono mescolare solo orbitali atomici di energie simili;
3) il numero di orbitali che si ottengono dopo l’ibridizzazione è uguale al numero di orbitali che si avevano prima;
4) il processo di ibridizzazione riguarda gli orbitali atomici e non gli elettroni: una volta ottenuti gli ibridi, in essi si sistemano gli elettroni in accordo con il principio di Pauli e con tutte le altre regole che conosciamo;
5) una volta ibridizzati gli orbitali atomici, non esistono più gli orbitali di partenza;
6) gli ibridi hanno forma simile tra di loro; le differenze principali riguardano le loro orientazioni nello spazio;
7) all’orientazione spaziale non contribuiscono orbitali atomici di tipo s (che, come sappiamo, hanno simmetria sferica) ma solo gli orbitali atomici che hanno caratteristiche direzionali (come i p, i d, … ). Gli orbitali s, ibridizzandosi con orbitali direzionali, fanno ingrassare l’orbitale direzionale;
8) se si mescolano tra loro due orbitali direzionali con direzioni lungo l’asse x e lungo l’asse y, si ottengono due ibridi che giacciono sul piano xy; se si mescolano tra loro due orbitali direzionali con direzioni lungo l’asse x e lungo l’asse z, si ottengono due ibridi che giacciono sul piano xz; … ; se si mescola un orbitale s con un orbitale direzionale x si ottengono due ibridi che giacciono sull’asse x; se si mescola un orbitale s con un orbitale direzionale y si ottengono due ibridi che giacciono sull’asse y; … .
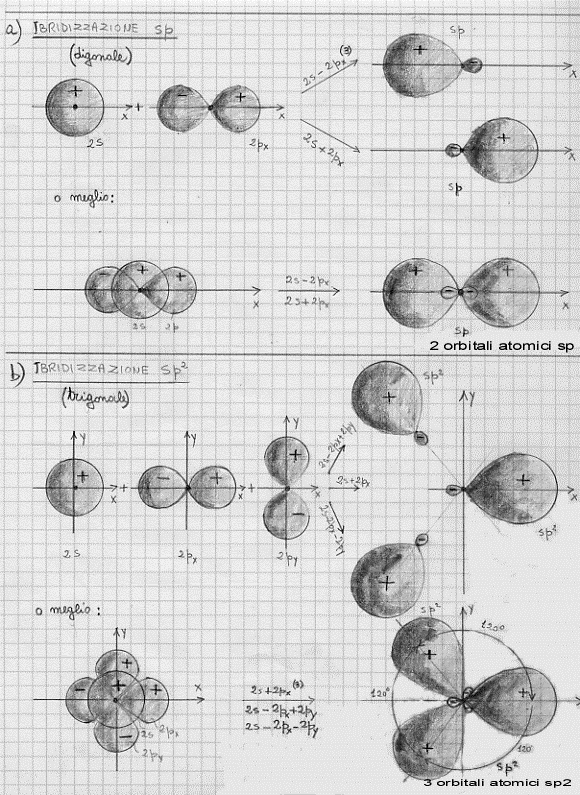
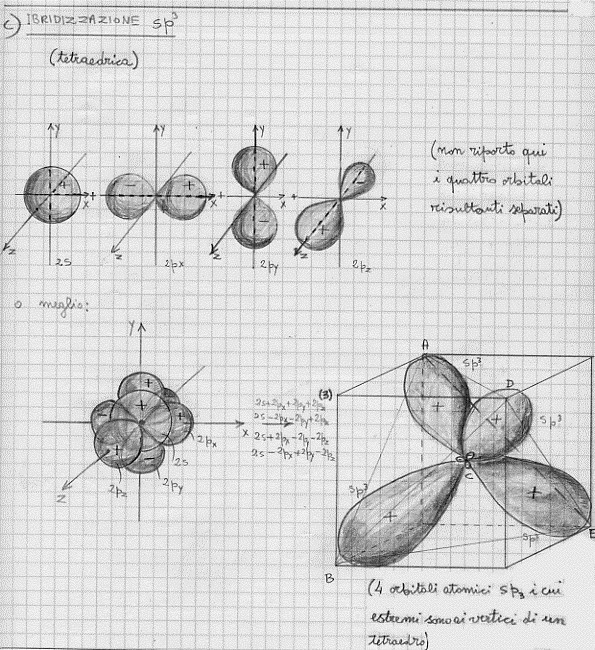
Figura 21
Le combinazioni di orbitali atomici indicate con (3) sono date a meno di alcuni fattori costanti detti di “normalizzazione”. Di questi fattori ne faremo sempre a meno.
Con questi nuovi orbitali atomici, gli ibridi, andiamo a vedere come si può rendere conto di differenti valenze e legami, andando in definitiva a studiare la struttura di qualche molecola poliatomica.
CH4 – molecola di metano (ibridi sp3)
Gli orbitali atomici che ci interessano sono gli orbitali ibridi sp3 che, come si può vedere dalla figura 21c, hanno la massima densità di carica elettronica in corrispondenza dei vertici del tetraedro ABDE nel quale l’atomo C di carbonio occupa il centro. D’altra parte sappiamo che l’idrogeno è descritto da uno stato 1s. E’ chiaro quindi che avvicinando 4 atomi di idrogeno ad un atomo di carbonio, poiché vale anche qui la regola della massima sovrapposizione,(15) essi tenderanno a sistemarsi proprio ai vertici del tetraedro ABDE, originando la classica configurazione tetraedrica della molecola CH4 di metano (figura 22):
C[(1s2)(2s2)(2px)(2py)] + 4H[(1s)] -> C[(1s2)(2s)(2px)(2py)(2pz)] + 4H[(1s)] -> C[(1s2)(sp3)4] + 4H[(1s)] -> CH4
C’è da notare che gli angoli a tra ogni coppia di assi unenti il nucleo del carbonio con i nuclei di idrogeno (figura 22a), sono tutti uguali e valgono 109° 28′, in perfetto accordo con le misure sperimentali eseguite con metodi spettroscopici.
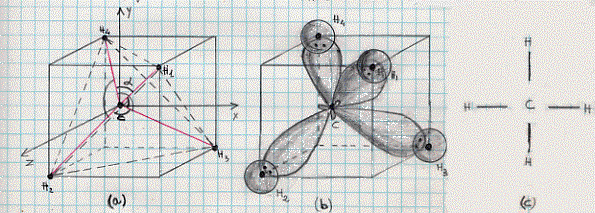
Figura 22
Infine occorre ricordare che altri elementi del gruppo del carbonio: come stagno, piombo, silicio e germanio, formano, anch’essi, una struttura tetraedrica intorno all’atomo centrale quando vanno a legarsi con altri elementi in svariati composti, ed inoltre gli angoli di legame risultano sempre di 109° 28′.
H2O – molecola d’acqua (ibridi sp3)
La molecola d’acqua è ancora lungi dall’essere compresa con una qualche precisione e si sono fatti vari tentativi per rendere conto del fatto sperimentale che più sfugge ad ogni descrizione teorica. Si è infatti osservato sperimentalmente che questa molecola presenta un angolo di legame di circa 105° (figura 23).
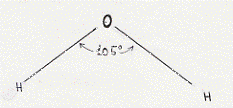
Figura 23
Utilizzando il metodo O.M. (si veda più oltre) si trova che quest’angolo dovrebbe essere di 90°; utilizzando gli ibridi sp3 quest’angolo diventerebbe di circa 109° che approssima meglio il dato sperimentale (tra le altre vi è poi stata una spiegazione mediante parziale ibridizzazione sp2).
La configurazione elettronica dell’ossigeno è:
O[(1s2)(2s2)(2px)(2py)(2pz2)]
e la prima cosa che può venire in mente riguarda il come sia possibile qui costruire ibridi sp3 .
Ebbene: si considerano gli orbitali atomici 2s, 2px , 2py , 2pz indipendentemente dagli elettroni che essi contengono e si costruiscono gli ibridi sp3 .Quindi si sistemano gli elettroni: una coppia in un sp3, un’altra coppia su un altro sp3 (queste coppie sono dette solitarie), un singolo elettrone su un sp3 ed un altro singolo sull’ultimo sp3 . Solo allora i due ibridi sp3 che contengono elettroni disaccoppiati, quelli che vanno a formare i due legami di tipo s (con lo stesso valore di energia), ad elettroni localizzati, con i due idrogeni (figura 24).
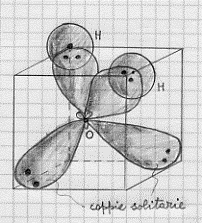
Figura 24
Ora, come abbiamo detto quando abbiamo trattato il legame idrogeno, le molecole d’acqua, nel liquido, non sembra si possano considerare isolate: pare che esse esistano in gruppi tenuti insieme, appunto, tramite il legame idrogeno. Questi gruppi sono formati da una molecola d’acqua circondata da altre molecole d’acqua che, quando arrivano a 4 (caso del ghiaccio), sono disposte ai vertici di un tetraedro (figura 25). Nel caso del ghiaccio vi sono allora più strutture
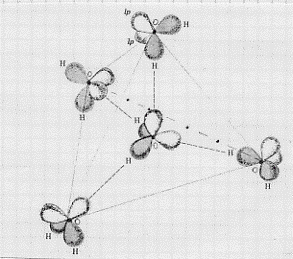
Figura 25 – I lobuli ombreggiati sono quelli che contengono coppie di elettroni leganti; gli altri contengono coppie solitarie. Le linee tratteggiate rappresentano il legame idrogeno. Le linee continue e quella tratto-punteggiata descrivono il tetraedro ai cui vertici si incontrane le quattro molecole di acqua che circondano la molecola centrale.
tetraedriche, come quella mostrata in figura 25, impacchettate tra loro in modo da formare una struttura complessiva come quella illustrata nelle figure 26 a e b (in b è mostrata una parte della struttura a da un altro angolo visuale). Come si può vedere la struttura è molto aperta (difficoltà di impacchettare tetraedri) e questo rende conto della bassa densità e quindi
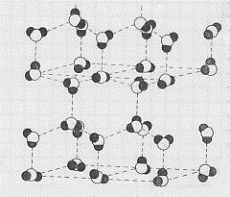
Figura 26 a
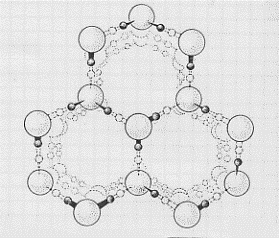
Figura 26 b
del maggiore volume occupato dal ghiaccio rispetto all’acqua.(16) Nel ghiaccio tutti i possibili legami idrogeno sono utilizzati; al crescere della temperatura sono sempre meno i legami idrogeno in azione e quindi via via va rompendosi la struttura cristallina; quando si ritrova acqua, ancora alcuni legami idrogeno sussistono(17) e questo rende conto del fatto che le molecole d’acqua si trovano in gruppi.
Come accennato, un’altra possibilità di spiegare i dati sperimentali per la molecola d’acqua è di considerare una parziale ibridizzazione sp (figura 21 b). Abbiamo già detto che legando tra loro due orbitali 2p puri (teoria O.M.) si avrebbe un angolo di 90°. Per arrivare ai circa 105° non basta considerare la repulsione tra i nuclei dell’idrogeno, poiché essa porterebbe l’angelo di legame ad un massimo di 95°;(18) bisogna invece ammettere che gli orbitali atomici 2p abbiano una parte di carattere s, bisogna cioè ammettere che si formino degli ibridi parziali sp2 . L’ introduzione di una componente s nei 2p permette una maggiore sovrapposizione e quindi un legame più stabile ed inoltre fornisce un angolo più vicino ai 105° di quanto non lo sia quello che si ricava dall’ibridizzazione sp3 .
NOTE
(1) Si tenga comunque conto che i due orbitali atomici, per formare un orbitale molecelare, debbono avere lo stesso numero quantico m.
(2) Si noti che, per quel che riguardava le molecole biatomiche omonucleari, il soddisfacimento della prima condizione implicava quello delle altre due. Ora invece le cose non stanno più così.
(3) Poiché qui avremo a che fare con combinazioni di orbitali atomici di tipo differente tra loro, ad evitare equivoci, useremo il simbolismo introdotto in nota 9 del precedente paragrafo.
(4) Si avrà sovrapposizione anche tra l’orbitale 1s dell’idrogeno e gli altri del fluoro, ma essa sarà minore. Calcoli più precisi dovrebbero tener conto anche di queste altre sovrapposizioni.
(5) L’orbitale molecolare di figura 18 b è di tipo s poiché ha una forma analoga agli orbitali molecolari di tipo s mostrati in figure 14 a e 14 b.
(6) Sperimentalmente si misura il momento di dipolo di una molecola e da esso si risale a k.
(7) L’elettronegatività, lo ricordo, è definita proprio da ciò che abbiamo detto. In modo più semplice si può dire che l’elettronegatività rappresenta il potere di un atomo di una molecola di attrarre elettroni. Si noti che il fluoro è il più elettronegativo tra gli elementi; esso è seguito dall’ossigeno, dal cloro e azoto, dal bromo, dal carbonio e zolfo, … .
(8) Tra i 2p vi è però solo il 2px che può dare un contributo al legame molecolare, poiché per i 2py e 2 pz vale quanto detto a proposito della figura 19.
(9) Si ricordi che il livello wp è doppiamente degenere e quindi in esso possono prendere posto 4 elettroni.
(10) Il procedimento di cercare analogie tra molecole costituite dallo stesso numero di elettroni (isoelettroniche) è stato criticato nel 1947 da Long e Walsh.
(11) Quando detto ha trovato una verifica in un gran numero di esperienze spettroscopiche.
(12) Pauling (1931), per spiegare la stabilità delle molecole con un numero dispari di elettroni (tra cui, appunto, NO), introdusse un nuovo tipo di legame, quello detto a tre elettroni. Con un metodo diverso si arriva allo stesso risultato che fornisce il metodo O.M.
(13) Gli orbitali di tipo p sono direzionali contrariamente agli orbitali di tipo s; ed i legami che si formano tra orbitali direzionali sonno più forti di quelli localizzati indifferentemente in una qualunque direzione.
(14) E’ indispensabile ricordare la differenza esistente tra livelli energetici e strati di livelli (K, L, M, …); e mentre tra strato e strato vi è una notevole differenza di energia, la differenza di energia esistente tra livello e livello all’interno di un medesimo strato è relativamente picco la. L’elio, in particolare, ha lo strato K pieno; mentre al berillio manca no 6 elettroni per completare lo strato L. All’interno dello strato L, proprio per le piccole differenze di energia esistenti tra i livelli ivi presenti, sono facilmente realizzabili transizioni di elettroni da un livello ad un altro. Ciò non accade per transizioni dallo strato K allo strato L (caso dell’elio).
(15) Si osservi che la sovrapposizione per un orbitale ibrido è sempre maggiore di quanto non accada per orbitali di tipo s o p puri.
(16) Le forme cristalline in cui può esistere il ghiaccio sono diverse (come hanno mostrato studi condotti sotto la direzione di Bridgmann e lavori di Lonsdale del 1958). Tutte le forme cristalline del ghiacci®o sono più dense dell’acqua, tranne il ghiaccio I che è meno denso proprio perché ha la struttura aperta (esagonale) mostrata in figura 26. A temperatura di – 80 °C il ghiaccio cristallizza nella forma cubica, come il diamante (si veda più avanti). E’ proprio il legame idrogeno che favorisce la struttura aperta del ghiaccio I. Quando si ha una prevalenza di questo legame vuol dire che le molecole sono disposte in modo tale da formare un cristallo con ampi spazi vuoti. Una diversa orientazione delle molecole non permetterebbe l’esistenza del legame idrogeno.
(17) Ancora a 40 °C vi sono circa la metà dei legami idrogeno possibili che ciascuna molecola d’acqua utilizza ancora (e ciò è in accordo con il fatto, sostenuto nel 1933 da Bernal e Fowler, che l’acqua manterrebbe una struttura a legami idrogeno simile a quella del ghiaccio).
(18) Come hanno mostrato Heath e Linnett nel 1948.
1 – Molecole poliatomiche trattate con il metodo L.V.: l’ibridizzazione (seconda parte)
BH3 – molecola di borano (1) (ibridi sp2)
Come abbiamo già visto, la molecola del boro può essere mutata mediante eccitazione:
B[(1s2)(2s2)(2px)] -> B[(1s2)(2s)(2px)(2py)] -> B[(1s2)(2sp2)3]
in tal modo si ottengono tre ibridi sp2 e l’atomo acquista caratteristiche di trivalenza. Risulta semplice quindi combinare l’atomo di boro con i suoi tre ibridi sp2 , ed i tre atomi di idrogeno (tenendo sempre conto della regola della massima sovrapposizione). Si ottiene così quanto mostrato in figura 27 (la a per una data orientazione degli assi e la b per una diversa orientazione). Il fatto poi che i legami formino tra loro angoli di 120° è confermato dalle misure sperimentali.
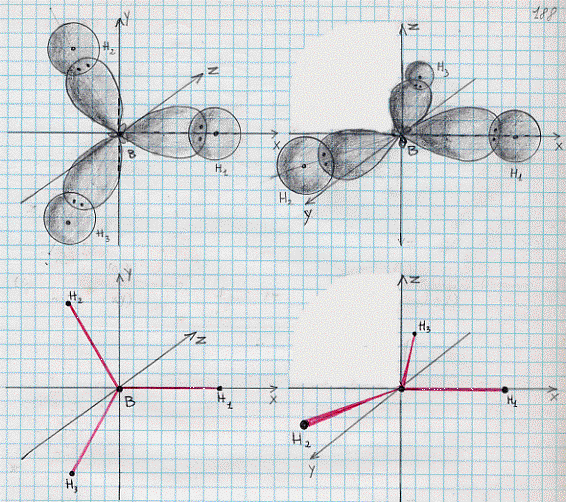
Figura 27
(a) I legami BH1, BH2 e BH3 si trovano sul piano xy.
(b) I legami BH1, BH2 e BH3 si trovano sul piano xy.
BF3 – molecola di trifluoruro di boro (ibridi sp2)
Nella discussione precedente abbiamo visto che per il boro si ha:
B[(1s2)(2sp2)3]
II fluoro ha invece la seguente configurazione elettronica (si è scelto il 2px parzialmente pieno, ma si poteva indifferentemente scegliere il 2py o il 2pz):
F[(1s2)(2s2)(2px)(2py2)(2pz2)]
e risulta quindi monovalente.
Con uno studio simile a quello fatto per la molecola di borano, possiamo arrivare a costruirci la molecola di trifluoruro di boro. I tre ibridi sp2 del boro si combineranno con i tre orbitali atomici 2px di tre atomi di fluoro in tre legami di tipo s, come mostrato in figura 28 (nella quale, come sempre, non abbiamo riportato gli altri orbitali non interessati al legame).(2) In questo modo si saturano le tre valenze del boro e tutto sembra a posto. Ma qui, ricordando
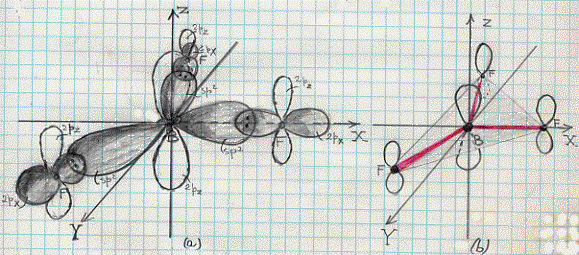
Figura 28 – Gli assi X, Y, Z, sono relativi al boro gli orbitali di tipo p del fluoro sono i 2px ; i legami si trovano sul piano XY del boro; gli orbitali non ombreggiati sono i 2pz del fluoro ed il 2pz del boro.
quanto abbiamo discusso a proposito della teoria O.M., c’è la possibilità di un ulteriore legame, anche se più debole: quelle tra gli orbitali 2pz del boro ed i tre orbitali 2pz dei tre atomi di fluoro (figura 29). Ricordando quanto sappiamo

Figura 29
sulla risonanza, si può dire che il legame (che sarà di tipo p) risuona tra i tre possibili, fornendo un’energia addizionale al legame stesso. Da quanto detto la molecola BF3 deve risultare molto più stabile della molecola BH3 e questo fatto è in perfetto accordo con le misure sperimentali. Così come è in accordo con i dati sperimentali il fatto che i 4 atomi che formano la molecola sono complanari, con angoli di legame di 120°.
C2 H4 – molecola di etilene (ibridi sp2) .
Ritorniamo ora ad un altro composto del carbonio, l’etilene, che ha molecola C2 H4. I dati sperimentali mostrano che in questa molecola tutti e sei gli atomi che la compongono giacciono su uno stesso piano e che gli angoli di legame sono di circa 120°. L’etilene presenta una struttura come quella mostrata in figura 30 e per spiegarla occorre ragionare
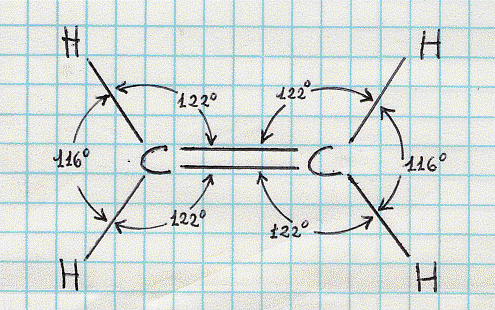
Figura 30
nel modo seguente. Come già sappiamo la configurazione elettronica del carbonio è:
C[(1s2)(2s2)(2px)(2py)] -> (eccitando) C[(1s2)(2s)(2px)(2py)(2pz)] -> C[(1s2)(sp2)3(2pz)].
Come si vede non abbiamo mescolato tutti e quattro gli orbitali 2s, 2px, 2py e 2pz per formare quattro ibridi sp3;(3) abbiamo ibridizzato solo tre orbitali e conseguentemente abbiamo ottenuto tre ibridi sp2 e ci è rimasto un orbitale atomico puro 2pz . E questo proprio per rendere cento dei fatti sperimentali, come cercheremo di mostrare.
Dalla figura 30 risulta che ciascun carbonio è tetravalente e che ogni carbonio, oltre a legarsi con un legame semplice con un idrogeno, si lega all’altro carbonio con un legame doppio.(4) I due atomi di carbonio possono allora venir separatamente descritti come in figura 31a.. Quando sono avvicinati (figura 31b) originano la struttura di figura 31c. Ed allora, oltre al legame di tipo s , che ha luogo direttamente tra i due atomi di carbonio per sovrapposizione
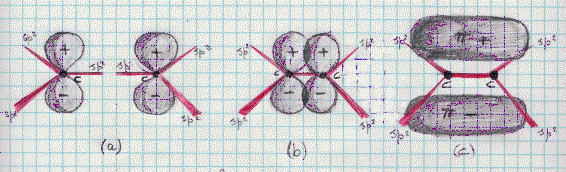
Figura 31
di due ibridi sp2 (si veda la figura 32), si ha a che fare anche con un legame p,(5) che conosciamo dalla teoria
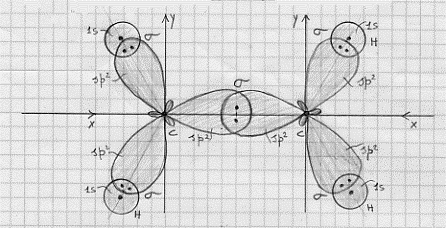
Figura 32
dell’orbitale molecolare (si noti che il piano della molecola, il suo scheletro, è, per l’orbitale p, un piano nodale). A questo proposito è importante ribadire quanto già detto: da un certo punto in poi, ed in particolare per le molecole poliatomiche, non c’è più distinzione tra la teoria O.M. e la teoria L.V.
Nella figura 32 sono illustrati: i 4 legami s dei 4 ibridi sp2 del carbonio con i 4 orbitali atomici 1s dell’idrogeno; l’unico legame s tra i due ibridi sp2 rimanenti dei due atomi di carbonio (in figura non è riportato il legame p).
C6H6 – molecola di benzene (ibridi sp2).
Sperimentalmente la molecola di benzene si presenta come un esagono regolare con angoli di legame di 120° (figura 33a). Dobbiamo quindi far ricorso agli ibridi sp2 del carbonio per costruire i legami s che costituiscono lo
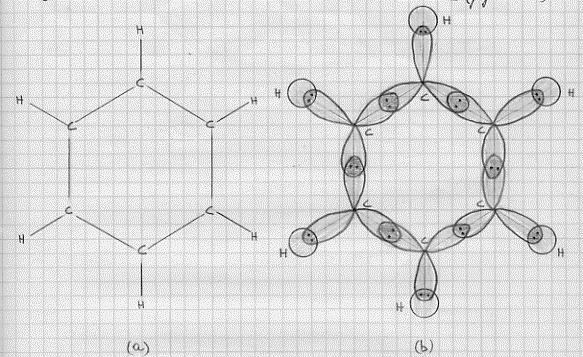
Figura 33
scheletro della molecola (figura 33b). Come si può vedere dalla figura si hanno 12 legami di tipo s che ci rendono ben conto della geometria della molecola osservata sperimentalmente. Bisogna ora prendere in considerazione i 6 orbitali atomici del carbonio di tipo 2pz che risultano perpendicolari al piano della figura 33. Indicando lo scheletro della molecola con un esagono, la situazione iniziale è mostrata in figura 34 (per brevità gli orbitali atomici 2pz sono stati
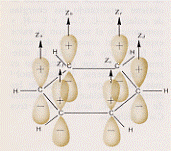
Figura 34
indicati con z1 , z2 , … ). Ora, su ciascun orbitale 2pz vi è un solo elettrone, di conseguenza, nella molecola, potranno formarsi tre legami molecolari p localizzati, ciascuno legando una coppia di elettroni. Ma quali orbitali atomici 2pz saranno, a coppie, interessati a questi legami ? Bisognerà tener conto di tutte le possibilità e quindi tener conto della risonanza tra le diverse strutture. Tutte le 5 possibilità di legame p, a coppie distinte di 2pz, sono riportate in figura 35.(6)
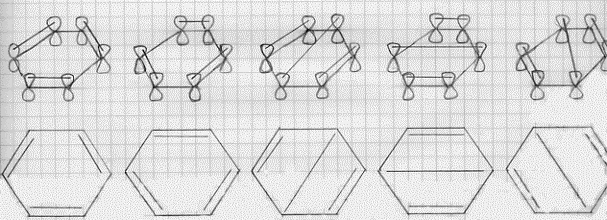
(a) (b) (c) (d) (e)
Figura 35
Si tratta allora di costruirsi una funzione d’onda che sia combinazione lineare di tutte le 5 strutture possibili; questa funzione d’onda è:
ψ = k1 ( ψa + ψb ) + k2 ( ψ c + ψd + ψe )
dove k1, e k2 sono due costanti da determinarsi e ψ a e ψb sono le due strutture che compaiono in figura 35 a e b (strutture di Kekulé), mentre ψ c , ψd e ψ e sono le tre strutture che compaiono in figura 35 c, d, e (strutture di Dewar). Si osservi che ragioni di simmetria fanno ammettere che le strutture di Kekulé e di Dewar hanno uguale peso nella costruzione della ψ .(7)
E da notare che è necessario costruirsi questa funzione d’onda, combinazione lineare di varie strutture, per rendere conto dei dati sperimentali che danno la molecola di benzene come più stabile di quello che sarebbe se si dovessero considerare solo sei legami semplici s tra atomi di carbonio e tre legami p ancora tra gli atomi di carbonio. Il valore addizionale dell’energia di legame che rende più stabile la molecola è fornito proprio dalla risonanza tra le diverse strutture, ognuna delle quali fornisce un proprio contributo a seconda del peso che ha nella funzione d’onda ψ.
BeH2 – molecola idruro di berillio(8) (ibridi sp)
II berillio ha la seguente configurazione elettronica:
Be[(1s2)(2s2)]
Eccitando opportunamente si ha:
Be[(1s2)(2s2)] -> (eccitando) Be[(1s2)(2s)(2px)] -> Be[(1s2)(sp)2].
Sono allora i due ibridi digonali sp (si veda la figura 21a) quelli che vanno a formare legami molecolari di tipo s con gli orbitali atomici 1s dell’idrogeno costruendo così la molecola di idruro di berillio (figura 36).
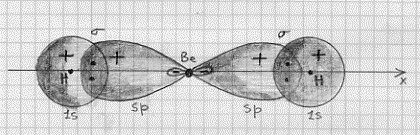
Figura 36
Come si vede la molecola presenta una geometria lineare, in perfetto accordo con i dati sperimentali. Il fatto poi che gli ibridi sp siano più allungati sull’asse x di quanto non lo fossero gli originali orbitali atomici 2px mostra un’altra possibile interpretazione del principio di Pauli: due elettroni con spin paralleli tendono a stare il più lontano possibile tra di loro.
C2H2 – molecola di acetilene (ibridi sp).
La molecola di acetilene, un altro importante composto del carbonio, risulta sperimentalmente lineare. Per rendere conto di ciò occorre ammettere che non si formino né ibridi sp3 né ibridi sp2 (sono solo gli ibridi sp che forniscono una perfetta simmetria intorno all’asse di legame e che, originando legami s, permettono la libera rotazione degli atomi costituenti la molecola intorno all’asse di legame). Inoltre la formula di struttura della molecola dovrà essere:
H —— C≡C——-H
dove ogni carbonio si lega con legame triplo con l’altro carbonio e con legame semplice con un atomo di idrogeno. In definitiva il carbonio risulta, in questa molecola, tetravalente e la configurazione elettronica che dobbiamo scegliere per esso è la seguente:
C[(1s2)(2s2)(2px)(2py)] -> (eccitando) C[(1s2)(2s)(2px)(2py)(2pz)] -> C[(1s2)(sp)2(2py)(2pz)]
Gli ibridi sp legheranno tra loro i due atomi di carbonio e questi ultimi con gli idrogeni mediante tre legami semplici s (figura 37); mentre gli orbitali atomici 2py dei due atomi di carbonio formeranno un orbitale molecolare p,
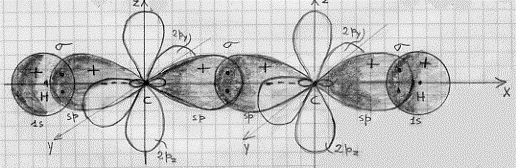
Figura 37 – Gli orbitali atomici non ombreggiati sono i 2py ed i 2pz dei due atomi di carbonio prima che si combinino per formare due orbitali molecolari p.
analogamente ai due orbitali atomici 2pz (figura 38).
Figura 38
Ed in ultima analisi la molecola ha una simmetria cilindrica intorno all’asse di legame (nel nostro caso l’asse x). I due atomi di carbonio sono legati tra loro con tre legami, uno semplice di tipo s e due di tipo p; se si aggiunge a questi l’ulteriore legame s che ogni atomo di carbonio ha con quello dell’idrogeno, si vede subito che è ben spiegata la tetravalenza del carbonio.
La molecola in oggetto ha una elevata energia di legame (più elevata dell’etilene e del metano) e questo fatto sembra confermare che gli ibridi sp si sovrappongono di più di quanto non facciano gli sp2 e gli sp3 ,con la conseguenza che ai legami sp corrisponde una maggiore energia, appunto, di legame.
CO2 – molecola di anidride carbonica (ibridi sp).
La configurazione elettronica del carbonio, nel caso si debbano considerare ibridi digonali sp, è:
C[(1s2)(sp)2(2py)(2pz)]
mentre per l’ossigeno possiamo dare, ad esempio, le due possibili seguenti:
O[(1s2)(2s2)(2px)(2py)(2pz2)] ; O[(1s2)(2s2)(2px)(2py2)(2pz)]
Per costruire la molecola si può pensare che i due ibridi sp del carbonio si leghino, lungo l’asse x, ciascuno con uno degli orbitali 2px dei due atomi di ossigeno (e questo fornisce le scheletro della molecola – mostrato in figura 39 – che, in accordo con i dati sperimentali, è lineare come del resto l’intera molecola). Consideriamo ora questo scheletro che,
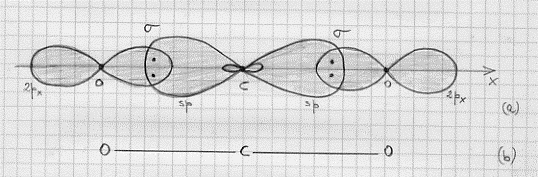
Figura 39 – La figura 39b non è ancora la formula di struttura ma solo lo scheletro della molecola.
come si vede dalla figura 39, è costituito da due legami semplici di tipo s; sovrapponiamo allo scheletro gli orbitali di tipo 2p, degli ossigeni e del carbonio, che ancora non abbiamo preso in considerazione (naturalmente non ci occupiamo degli orbitali 1s e 2s degli ossigeni e dell’1s del carbonio): l’atomo di carbonio ha due orbitali, 2py e 2pz , ambedue con elettroni disaccoppiati; gli atomi di ossigeno hanno un orbitale 2py (o 2pz ) completamente pieno ed un orbitale 2pz (o 2py ) con un solo elettrone. Riportiamo questa situazione al di sopra dello scheletro della molecola disegnando a tratto continuo gli orbitali pieni e tratteggiati quelli con un solo elettrone (figura 40). Osservando la figura si può vedere che abbiamo disegnato una delle due situazioni possibili. In figura, il primo ossigeno alla sinistra ha l’orbitale 2py pieno ed
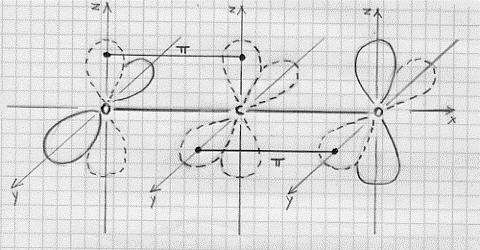
Figura 40
il 2pz semipieno, mentre l’altro ossigeno ha l’orbitale 2pz pieno ed il 2py semipieno. L’altra situazione possibile prevederebbe l’eventualità inversa. Bisognerà pertanto tenere conto della risonanza tra queste due possibili strutture.
In ogni caso, riferendoci alla situazione di figura 40, mentre il 2pz del primo ossigeno forma un legame p con il 2pz del carbonio, il 2py del secondo ossigeno forma un altro legame p con il 2py del carbonio (l’altra situazione è la simmetrica di quella descritta, tenendo in conto che ambedue portano alla stessa situazione energetica e pertanto, nella funzione d’onda che descrive la molecola CO2 , le due situazioni avranno uguale peso).
La formula di struttura dell’anidride carbonica sarà allora quella mostrata in figura 41a. A questo punto però vi
Figura 41
sono altre due possibili strutture da dover considerare (figura 41 b e c) e naturalmente un calcolo più completo dovrebbe prevedere la risonanza tra queste ultime tre strutture.
NOTE
(1) E’ una molecola che si osserva solo in particolari condizioni, quando si frammenta la molecola B2H6 di diborano.
(2) Si noti che gli assi X, Y, Z del boro non devono necessariamente avere le stesse orientazioni degli assi x, y, z del fluoro.
(3) Si deve scartare il fatto che si formino 4 ibridi sp3 perché in questo caso la molecola non sarebbe più piana e perché non sarebbe rispettata la regola della massima sovrapposizione. Questa teoria fu proposta da Slater e Pauling nel 1931; quella che illustriamo nel testo fu proposta da Huckel nel 1930 e sviluppata da Penney nel 1934.
(4) Quando si parla di legame doppio si intende un legame di tipo s ed un legame di tipo p. Altrimenti, se ci si vuol riferire a due legami di tipo s si parla di due legami semplici.
Si noti che sperimentalmente si osserva che, nella molecola di acetilene, i due frammenti molecolari CH2 non possono ruotare l’uno rispetto all’altro, come accadrebbe se si avesse a che fare con un legame semplice di tipo s ; questo è uno dei fatti che induce a pensare all’esistenza di un altro tipo di legame, quello p .
(5) Gli orbitali p di cui si tratta, si trovano localizzati tra i due atomi di carbonio (questa precisazione è utile per quanto vedremo più avanti a proposito di orbitali delocalizzati.
Si ricordi che un legame p ha energia minore di un legame s.
(6) II fatto che le possibili strutture sono 5 e che tutte le altre si possono far risalire a queste 5, fu mostrato nel 1935 da Van Vleck e Sherman. Riguardo poi le 5 strutture riportate, le prime due furono proposte da Kekulé nel 1865 e le ultime tre da Dewar, che per un poco di tempo fu suo allievo, qualche anno dopo.
(7) Anche se, in realtà, è stato mostrato che ogni struttura di Kekulé ha un peso del 39% mentre ogni struttura di Dewar ha un peso del 7%, fatto che indica la maggiore importanza delle strutture di Kekulé rispetto a quelle di Dewar (relativamente al contributo fornito all’energia di legame della molecola).
(8) L’idrogeno molecolare reagisce con il berillio solo ad alte temperature, formando la molecola BeH2.
1 – Molecole poliatomiche trattate con il metodo O.M.: orbitali molecolari localizzati e delocalizzati
Fino ad ora, in tutte le molecole delle quali ci siamo occupati, abbiamo fatto esplicito riferimento ad un tipo di legame che prevedeva la localizzazione di una coppia di elettroni, e quindi dell’orbitale molecolare che li rappresentava, in una zona bea precisa di spazio, compresa al massimo tra due nuclei. Più in generale, elettroni che siano confinati in un orbitale atomico di un atomo o nella regione di legame tra due atomi in un orbitale molecolare (soprattutto negli orbitali di tipo s più volte incontrati), originano orbitali localizzati.
La teoria L.V. usa esclusivamente di orbitali localizzati oltre al fondamentale concetto di risonanza(1) al quale, come abbiamo visto, corrisponde una quantità di energia di legame dipendente dal numero di strutture tra le quali la molecola risuona.
Anche la teoria O.M. si serve di orbitali molecolari localizzati, ed anzi quando ciò è possibile è preferibile, ma spesso si ha a che fare con sistemi per i quali è difficile mettere insieme una coppia di elettroni in un orbitale molecolare unico e quindi localizzato. In questo caso si deve introdurre il concetto di orbitali (molecolari) delocalizzati, l’analogo nella teoria O.M. di ciò che rappresenta la risonanza nella teoria L.V. (come dovrebbe essere ormai chiaro la teoria O.M. non si serve della risonanza). Quando un orbitale molecolare non è più un semplice orbitale molecolare, così come lo abbiamo descritto nelle pagine precedenti, ma è il risultato di una mescolanza tra orbitali molecolari ed atomici(2) allora esso diventa un orbitale molecolare delocalizzato.
E’ chiaro da quanto appena eletto che su un orbitale delocalizzato vi possono essere più di due elettroni e, poiché questo orbitale è sempre il risultato del legame che si forma tra più di due atomi, esso sarà un orbitale che interessa l’intera molecola, estendendosi al di sopra (e al di sotto) di tutti i nuclei che ne fanno parte. Così intendendo le cose, non avrà più senso dire che una coppia di elettroni è localizzata tra due nuclei; poiché l’orbitale delocalizzato si estende su tutti i nuclei, gli elettroni che in esso si trovano possono essere situati in qualunque zona dell’orbitale (che avrà simmetria di tipo p, con i nuclei giacenti nel piano nodale), conseguentemente in qualsiasi zona della molecola (all’interno dell’orbitale) ed in definitiva sono liberi di muoversi in tutta la molecola (all’interno dell’orbitale). A questo libero movimento degli elettroni nell’orbitale vi è una sola limitazione ed è ancora dovuta al principio di Pauli: nelle vicinanze di un nucleo, su un dato livello energetico di quell’atomo, non possono esservi più di due elettroni e questo fatto limita le possibili posizioni degli elettroni nell’intera molecola.
E’ interessante osservare che alla delocalizzazione degli orbitali corrisponde un’energia di legame, detta energia di delocalizzazione, che, facendosi i conti, ha lo stesso valore dell’energia di risonanza della teoria L.V.
Alcune volte, anche nel caso di molecole poliatomiche, sarà possibile darne una descrizione con la teoria O.M. utilizzando orbitali molecolari localizzati; altre volte ciò è praticamente impossibile, come nel caso degli stati eccitati di molecole contenenti non più di un doppio legame ed in quello delle molecole aromatiche (ad esempio: il benzene C6H6) e coniugate (3) (ad esempio: il butadiene C4H6), e bisognerà ricorrere agli O.M. delocalizzati.
Di seguito vedremo degli esempi di un caso e dell’altro, trattando di varie molecole poliatomiche (già trattate con il metodo L.V.: proprio per porre in evidenza le differenze di trattazione tra i due metodi), e dagli esempi risulterà anche più chiaro quanto abbiamo fin qui detto a proposito di O.M. localizzati e delocalizzati.
Naturalmente si opererà anche qui in approssimazione L.C.A.O.
BeH2 – molecola di idruro di berillio. (O .M. delocalizzati)
Questa, semplice molecola poliatomica, che già abbiamo visto essere lineare, ci offrirà lo spunto per vedere come il metodo O.M. vada applicato a molecole con più di due atomi.(4)
Cominciamo con il ricordare la configurazione elettronica del berillio:
Be[(1s2)(2s2)] -> (eccitando) Be[(1s2)(2s)(2px)];
gli orbitali di valenza del berillio sono quindi il 2s ed il 2px, mentre l’idrogeno ha l’orbitale di valenza 1s. Si tratterà quindi di combinare, in orbitali molecolari, gli orbitali atomici 2s e 2px del berillio con i due orbitali atomici 1s dei due idrogeni (che per comodità indichiamo con Ha ed Hb). Si devono cioè trovare delle funzioni d’onda molecolari che siano opportune combinazioni lineari delle funzioni d’onda atomiche e nel far ciò è molto importante tener conto del segno che le funzioni d’onda atomiche (o gli orbitali atomici) hanno. Con un disegno possiamo capire meglio: serviamoci quindi di figura 42. La figura 42a mostra, la sovrapposizione dell’orbitale 2s del berillio con gli orbitali 1s
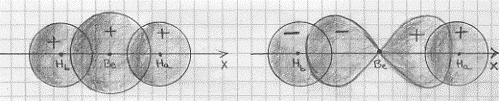
Figura 42
degli idrogeni. Come si può vedere si hanno sempre gli stessi segni per gli orbitali atomici e, conseguentemente, la frazione d’onda molecolare (legante) che rappresenterà questa situazione sarà, a meno di fattori moltiplicativi costanti detti di normalizzazione, la semplice somma delle singole funzioni d’onda atomiche:
(1) ψ (s2s)= ψ Hb (1s) + ψBe (2s) + ψ Ha (1s)
La figura 42b mostra la sovrapposizione dell’orbitale 2px del berillio con gli orbitali 1s degli idrogeni. Qui ci troviamo di fronte ad una situazione differente da quella appena vista. Ora il 2px del berillio ha un lobo positivo ed uno negativo mentre gli 1s degli idrogeni sono sempre positivi. Per poter fare una combinazione lineare di questi orbitali atomici per ottenere un orbitale molecolare legante, occorrerà cambiare segno all’orbitale atomico 1s dell’idrogeno Hb(5) (a sinistra in figura 42b) e, per farlo, basta cambiare segno alla funzione d’onda che lo rappresenta. In questo caso, sempre a meno di fattori moltiplicativi costanti o di fattori di normalizzazione, la funzione d’onda molecolare (l’orbitale molecolare) legante sarà:
(2) ψ (s2px)~ ψ Ha (1s) + ψBe (2px) – ψ Hb (1s)
Con un analogo ragionamento siamo in grado di costruirci gli orbitali molecolari antileganti s*. Si tratta di fare una sovrapposizione come quella mostrata in figura 42, solo che ora occorrerà cambiare i segni degli 1s degli idrogeni rispetto a quelli indicati nella figura 42. Si ha allora:
(3) ψ (s*2s)~ – ψ Hb (1s) + ψBe (2s) – ψ Ha (1s)
(4) ψ (s*2px)~ – ψ Ha (1s) + ψBe (2px) + ψ Hb (1s)
Ricordando ora quanto abbiamo detto a proposito delle combinazioni lineari è più corretto scrivere le (l), (2) ,(3) e (4) nel modo seguente:
ψ (s2s)= ψ Be (2s) + k1[ψHa (1s) + ψ Hb (1s)]
ψ (s2px)~ ψ Be (2px) + k2[ψHa (1s) – ψ Hb (1s)]
ψ (s*2s)~ ψ Be (2s) – k3[ψHa (1s) + ψ Hb (1s)]
ψ (s*2px)~ ψ Be (2px) – k4[ ψBe (1s) – ψ Hb (1s)]
dove k1 , k2 , k3 e k4 sono, al solito, delle costasti che ci danno indicazioni sul peso relativo degli orbitali atomici di valenza del berillio e dell’ idrogeno nella formazione della molecola; esse vanno determinate a partire dalla condizione che l’energia deve risultare minima, affinché il legame sia stabile. Facendo i conti, che sono al di fuori della nostra portata, si trova che le superfici limite degli orbitali molecolari s e s*, per la molecola di BeH2 , sono quelli riportati in figura 43. Inoltre, poiché risulta che gli orbitali molecolari più stabili sono quelli leganti, i 4 elettroni di valenza della molecola di BeH2 si sistemeranno in essi e, in definitiva, la configurazione elettronica della molecola sarà:
BeH2[k(s2s)2(s2px)2]
e, come si vede, le due coppie di elettroni che formano il legame sono delocalizzati sui tre atomi che formano la molecola (non c’è modo di localizzare le coppie di elettroni).
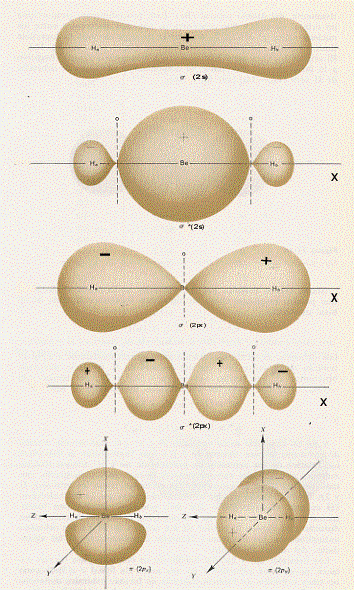
Figura 43
CO2 – Molecola di anidride carbonica (O.M. delocalizzati)
Ricordiamo le configurazioni elettroniche del carbonio e dell’ossigeno:
C[(1s2)(2s2)(2px)(2py)] ; O[(1s2)(2s2)(2px)(2py)(2pz2)]
Gli orbitali di valenza del carbonio sono il 2s , il 2px ed il 2py ; quelli dell’ossigeno sono il 2px , il 2py ed il 2pz (avendo trascurato per semplicità il 2s2 ). Come si ricorderà anche questa è una molecola lineare.
La sovrapposizione degli orbitali 2s e 2px del carbonio con i due orbitali 2px dei due ossigeni ci forniranno i legami di tipo s e s*; la sovrapposizione degli orbitali 2py e 2pz(6) dei tre atomi ci fornirà i legami di tipo p e p* . Le combinazioni lineari degli orbitali atomici che ci forniscono gli orbitali molecolari saranno allora:(7)

Le combinazioni lineari cosi ottenute sono in numero di 10. Per le prime 8 non c’è altro da fare che la figura che ne rappresenta le superfici limite (figura 44), poiché la formazione degli orbitali molecolari avviene allo stesso modo che abbiamo visto nel caso di BeH2, con la sola differenza che ora sono i 2px degli ossigeni anziché gli 1s degli idrogeni a formare orbitali molecolari s e s*. Le ultime due combinazioni lineari abbisognano invece di una spiegazione.
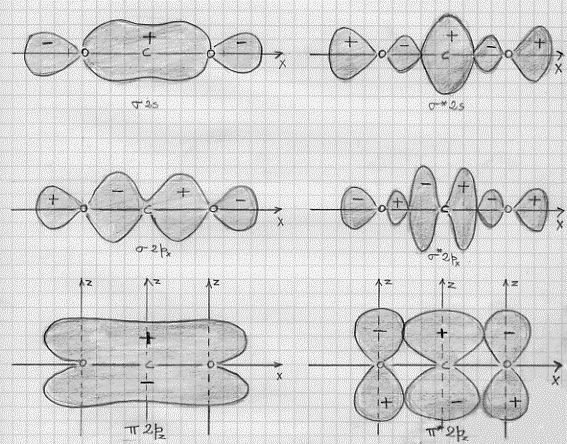
Figura 44 – Non sono stati disegnati i p2py e p*2py perché equivalenti, rispettivamente, ai p2pz e p*2pz .
Discuteremo solo la prima delle due, poiché per l’altra vale un discorso esattamente equivalente, servendoci della figura 45a nella quale sono rappresentati gli orbitali atomici 2pz di ciascuno dei tre atomi che vanno a formare la molecola. La figura corrisponde alla combinazione lineare:
y (p2pz)= y C (2pz) + k7[ yO (2pza) – y O (2pzb)]
e, come si può vedere dalla figura, tra i vari 2pz non vi è una sovrapposizione netta a causa del segno di 2pzb che,
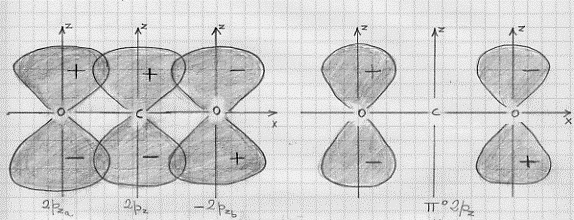
Figura 45
essendo negativo, non permette che si abbia una y interamente positiva al di sopra dell’asse x ed interamente negativa al di sotto del medesimo asse. Di conseguenza la cosa va come se non vi fosse alcuna sovrapposizione con l’orbitale 2pz del carbonio e si dovessero considerare solo i 2pz degli ossigeni. Rimane allora solo la combinazione 2pza – 2pzb che equivale a porre y C (2pz) = 0 nella relazione precedentemente scritta; e questa combinazione è anch’essa un orbitale molecolare costituito da due orbitali atomici che praticamente non interagiscono tra di loro per la distanza a cui si trovano. E se non c’è interazione, non c’è cambio nel valore dell’energia (si riveda la figura 12) che resta quello che compete agli orbitali atomici dei due ossigeni. In questo modo l’orbitale molecolare risultante non è né legante né antilegante: esso è detto non legante ed indicato ,con il simbolo p°2pz (si veda figura 45b).
In definitiva, la configurazione elettronica di CO2 è data da:
CO2[KK(2sa2)(2sb2)(s2s)2(s2px)2(p2py)2(p2pz)2(p°2py)2(p°2pz)2],
ed anche qui gli orbitali molecolari risultano completamente delocalizzati. Si deve notare che l’energia di p2py è la stessa di p2pz, che l’energia di p°2py è la stessa di p°2pz che come al solito gli altri valori dell’energia sono scritti in successione crescente e che non vi sono elettroni in orbitali antileganti.
BF3 – molecola di trifluoruro di boro (O.M. delocalizzati)
Iniziamo con il ricordare che questa molecola ha una struttura piana trigonale e che le configurazioni elettroniche del boro e del fluoro sono:
B[(1s2)(2s2)(2px)] ; F[(1s2)(2s2)(2px)(2py2)(2pz2)]
saranno quindi gli orbitali atomici 2s e 2px del boro a legarsi in orbitali molecolari con gli orbitali atomici 2s e 2px del fluoro. Ricordiamo anche che, se pure non compaiono nella configurazione elettronica del boro, occorrerà prendere in considerazione anche i suoi orbitali atomici 2py e 2pz che, come sappiamo, sono vuoti. Poiché poi abbiamo a che fare con tre atomi di fluoro, ci serviremo dei subindici a, b, c per indicarli. Infine dobbiamo osservare che le coordinate x, y, z per gli orbitali atomici del boro non coincidono con quelle del fluoro (proprio perché la molecola è trigonale piana).
Fatte queste premesse, iniziamo con il disegnarci queste sovrapposizioni e quindi a costruirci gli orbitali molecolari s, dovuti alla sovrapposizione degli orbitali atomici 2s, 2py e 2pz del boro con gli orbitali 2pxa , 2pxb , 2pxc dei fluori, ed infine i p, dovuti alla sovrapposizione del 2pz del boro e dei 2py dei fluori. Nella figura 46 sono riportate:
in (a) le sovrapposizioni del 2s del boro con i 2px dei fluori;
in (b) quelle del 2py del boro con i 2px dei fluori;
in (c) quelle del 2px del boro con i 2px dei fluori;
in (d) quelle del 2pz del boro con i 2py dei fluori;
in (e) ed in (f) due possibili orbitali non leganti che anche in questo caso si producono e che discuteremo tra poco.
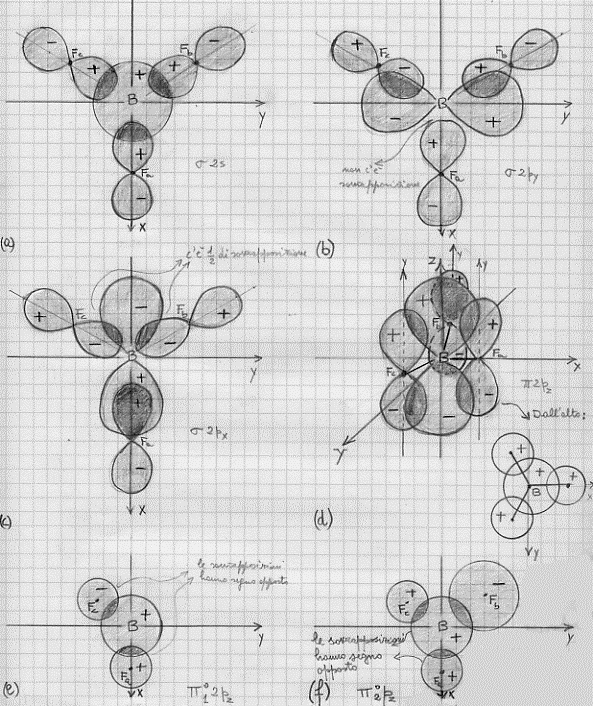
Figura 46
Le funzioni d’onda molecolari, combinazioni lineari delle funzioni d’onda atomiche, che ci rappresentano ordinatamente le situazioni di figura 46, sono:
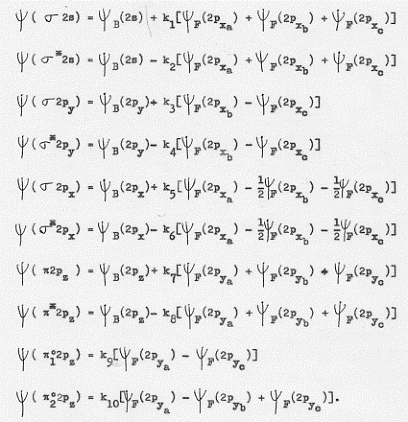
(Qui, come anche nella fig. 46, compare uno ‘strano’ orbitale molecolare s2py. Ebbene esso non ha nulla a che vedere con gli orbitali molecolari definiti in figura 14, tanto più che questo tipo di orbitale non esisteva proprio – gli orbitali di tipo s potevano essere realizzati solo dalla sovrapposizione di orbitali atomici di tipo s o px -. Gli orbitali molecolari dei quali discutiamo ora (caso di molecole poliatomiche) sono originati da diverse sovrapposizioni tra orbitali atomici di tipo differente ed essi saranno s o p, a seconda della forma risultante).
Non resta ora che dare la configurazione elettronica di BF3:
BF3[KK(2sa2)(2sb2)(s2s)2(s2py)2(s2px)2(p2pz)2(p°12pz)2(p°22pz)2(2pza2)(2pzb2)(2pzc2)],
come si vede, vi sono 6 elettroni in orbitali leganti s e ciò vuol dire che si hanno 3 legami s; vi sono inoltre due elettroni in un orbitale legante p che formano un legane p che risulta completamente delocalizzato (i due elettroni che formano il legame p appartengono all’intera molecola). Si può rappresentare questo legame p delocalizzato come in figura 47.
Figura 47
H2O – molecola d’acqua (O.M. delocalizzati)
Senza entrare in troppi dettagli, si tratterà di costruire delle combinazioni lineari degli orbitali atomici 2s, 2px , 2py dell’ossigeno con gli 1s degli idrogeni. Nel far questo dovremo tener conto che sperimentalmente l’angolo di legame risulta di circa 105°.
Le possibili sovrapposizioni (combinazioni lineari) leganti sono mostrate in figura 48. Si deve però tener conto che le ultime due sovrapposizioni, s2px e s2s (figura 48b e 48c), avendo la stessa simmetria, si combinano tra di loro in una specie di ibridizzazione per formare un orbitale molecolare fortemente legante (s2s) ed uno con carattere parzialmente non legante, che chiameremo s x. Vi sono poi i due elettroni dell’ossigeno che si trovano sull’orbitale atomico 2pz; ma quest’ultimo non ha alcuna sovrapposizione con gli 1s degli idrogeni e pertanto si dovrà considerare un legame di tipo p non legante e cioè p°2pz . Infine si avranno gli orbitali molecolari non leganti (che si ottengono dalle
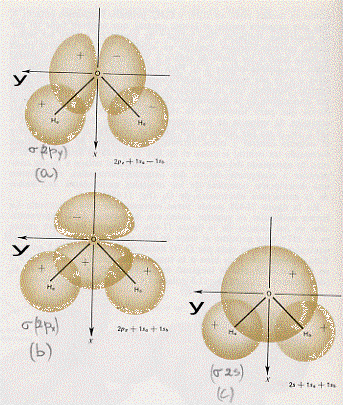
Figura 48
combinazioni mostrate in figura 48 quando si cambi segno agli ultimi tre termini) anche se su di essi non vi sono elettroni.
La configurazione elettronica di H2O sarà allora:
H2O[K(s2s)2(s2py)2(sx)2(p°2pz)2],
e, come si vede, si avranno due legami netti di tipo s. Si deve però osservare che se si avessero solo questi due legami netti l’angolo di legame sarebbe di 90°. E’ proprio l’ulteriore parziale legame (il sx), come hanno mostrato recentemente Pitzer e Cusachs, che fa si che l’angolo di legame tenda ai quasi 105°. Da questa discussione risulta che 6 elettroni sono in orbitali molecolari delocalizzati nell’intera molecola mentre solo due (i p°2pz) risultano localizzati. Infine, questa descrizione è in accordo con le misure spettroscopiche poiché tutti e quattro gli orbitali risultanti hanno energie distinte (nessun orbitale è degenere – si veda figura 49), contrariamente a quanto accadeva nel caso della stessa molecola discussa con il metodo L.V. e mediante ibridizzazione sp (allora si avevano due orbitali molecolari leganti s equivalenti in energia) e ciò vuol dire che è il metodo L.V. quello in accordo con l’esperienza spettroscopica.

Figura 49 – Si osservi che solo i primi 4 orbitali molecolari risultano pieni; quelli antileganti non contengono elettroni.
H2O – molecola d’acqua (O.M. localizzati)
Oltre alla trattazione della molecola d’acqua mediante gli orbitali molecolari delocalizzati, è possibile farne un’altra mediante l’uso degli orbitali molecolari localizzati. In questo caso occorrerà considerare la combinazione lineare degli orbitali atomici 1s degli idrogeni e degli orbitali 2px e 2py dell’ossigeno. Si può subito procedere ad una approssimazione (per altri versi molto ragionevole) che prevede separatamente la combinazione lineare dell’1s di un idrogeno con il 2px dell’ossigeno e dell’1s dell’altro idrogeno con il 2py dell’ossigeno.
Ricordando però che gli orbitali atomici 2px e 2py sono perpendicolari tra di loro ci si dovrebbe aspettare un angolo di legame di 90° (si veda figura 50 a), contrariamente al fatto, che già conosciamo e verificato sperimentalmente, che l’angolo di legame nell’acqua è di circa 105°. Per rendere conto di questa differenza (figura 50 b) si sono ricercate varie spiegazioni, tra le quali:
1) la repulsione elettrostatica tra gli atomi di idrogeno (elettrone-elettrone e protone-protone) che sono legati con l’ossigeno;
2) la repulsione tra i due legami O-H;
3) un particolare intervento delle coppie solitarie di elettroni (si riveda quanto abbiamo detto a proposito della molecola
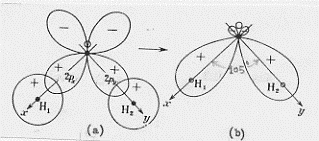
Figura 50
d’acqua quando la abbiamo trattata con il metodo L.V. mediante gli orbitali ibridi).
Un ultima cosa resta da dire a proposito della molecola d’acqua, almeno nell’ambito dei nostri scopi, ed è relativa al suo essere polare, al fatto cioè che ad essa si deve assegnare un momento di dipolo (md). Questo momento di dipolo è dovuto alla somma vettoriale del momento di dipolo originato dagli elettroni che formano il legame e del momento di dipolo originato dalle coppie di elettroni solitari. E mentre quest’ultimo è un unico momento di dipolo, l’altro è originato per somma vettoriale dei singoli momenti di dipolo di ciascun legame (si veda la figura 51).
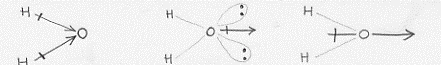
Dipoli di legame Dipolo di coppie solitarie Dipolo totale
Figura 51
C6H6 – molecola di benzene (O.M. delocalizzati )
La costruzione di questa molecola con la teoria degli orbitali molecolari si fa utilizzando in parte la teoria degli ibridi introdotta nel metodo L.V. ed in parte la teoria degli O.M. delocalizzati.
I 6 legami carbonio-carbonio ed i 6 legami carbonio-idrogeno si costruiscono esattamente nello stesso modo già discusso nel metodo L.V., mediante gli ibridi sp2 del carbonio e gli orbitali atomici 1s degli idrogeni. Questo procedimento ci fornisce i legami localizzati s che costituiscono lo scheletro della molecola (si riveda la figura 33). Restano ora da considerare i sei orbitali atomici 2pz dei sei atomi di carbonio (si riveda la figura 34) che ci forniranno i legami delocalizzati di tipo p. In effetti non vi è ragione di considerare una sovrapposizione piuttosto che un’altra di questi orbitali 2pz . Sembra lecito pensare ad una sovrapposizione cumulativa in modo da ottenere orbitali molecolari delocalizzati sull’intera molecola. E, dato che si hanno 6 orbitali atomici, si avranno 6 orbitali molecolari, 3 dei quali leganti e 3 antileganti. I 6 elettroni disponibili si troveranno sugli orbitali molecolari leganti che sono mostrati nella figura 52. Questi elettroni, liberi di muoversi nell’intera molecola, sono stati chiamati da Lennard-Jones (1937) elettroni mobili e Coulson ha proposto di considerarli come una piccola corrente elettrica che gira intorno all’anello e, poiché
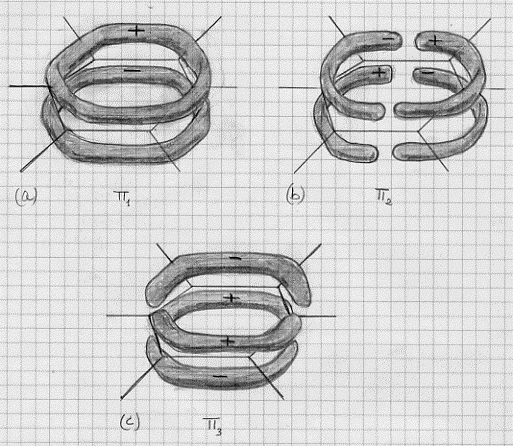
Figura 52
normalmente vi sono correnti uguali ruotanti in versi opposti, l’effetto totale sarà nullo. Ma si comincia qui a profilare un’analogia tra gli elettroni delocalizzati nel benzene e gli elettroni di conduzione nei metalli: in ambedue i casi una perturbazione elettrica provocata in una parte del sistema si propaga con facilità in altre parti.
II fatto poi che gli elettroni abbiano una zona più ampia in cui muoversi fornirà un valore più basso per l’energia totale e quindi una più alta energia di legame rispetto al caso degli orbitali localizzati; si tratta proprio di quell’energia delocalizzazione (Coulson, 1947) che è l’analogo O.M. dell’energia di risonanza L.V.
– La grafite ed il diamante –
E’ ora interessante almeno un cenno a queste due forme di cristallizzazione del carbonio. E’ istruttivo andare a vedere, per differenza tra grafite e diamante, quale ruolo giocano gli orbitali molecolari delocalizzati.
Iniziamo con la figura 53 che mostra la differenza tra le due strutture cristalline. Come si vede (figura 53b) la grafite, una delle forme solide nelle quali incontriamo il carbonio, ha una struttura cristallina a celle esagonali. Il
(a)
(b)
(c)
Figura 53
carbonio, tetravalente, si lega ai tre atomi di carbonio che gli giacciono vicini nello stesso piano mediante legami s dovuti ad ibridizzazione trigonale sp . In questo modo si saturano 3 valenze. L’altro legame è dovuto agli orbitali atomici 2pz del carbonio (che risultano perpendicolari al piano di figura 53b). Questo legame è di tipo p ed è completamente delocalizzato su tutto il piano degli esagoni. Ogni stato di esagoni costituisce usa lamina (una gigantesca molecola) che è legata alla lamina sovrapposta e sottoposta da deboli forze di Van der Waals. Si hanno in definitiva tante lamine sovrapposte (figura 53a): all’interno delle lamine vi sono forze intense contrariamente a ciò che avviene tra lamina e lamina. Quest’ultimo fatto spiega bene il potere lubrificante della grafite: sono le lamine che, scorrendo le une sulle altre, originano il fenomeno. Inoltre gli orbitali delocalizzati rendono conto del potere conduttore che questo materiale presenta.
Nel caso del diamante (figura 53c) quanto ora detto non si verifica poiché si ha cristallizzazione nella struttura tetraedrica a coppie di elettroni localizzati, dovuta ad ibridizzazione tetragonale sp3 che, come sappiamo, non lascia orbitali non mescolati. Questo tipo di cristallizazione, oltre a quante detto, rende conto della grande durezza del diamante e del suo potere isolante.
NOTE
(1) Approfitto dell’occasione per ricordare che se due (o più) sono le possibili strutture che noi utilizziamo per rappresentare una molecola, parlare di risonanza tra queste strutture significa solo dire che nessuna delle strutture rappresenta bene la situazione, la quale invece è meglio descritta dalla sovrapposizione (matematica) di tutte le possibili strutture (che sono solo dei modelli matematici senza alcuna realtà fisica).
(2) Si noti la differenza con gli ibridi: in quel caso si aveva mescolanza tra orbitali atomici e basta. Successi vamente questi nuovi orbitali atomici, gli ibridi, andavano a formare legami con altri orbitali atomici.
(3) Le molecole aromatiche sono quelle i cui atomi componenti sono disposti ad anello chiuso. Si hanno invece molecole coniugate quando in esse sono presenti due doppi legami separati da un legame semplice; ad esempio il butadiene ha la seguente formula di struttura: CH2=CH—CH=CH2 .
(4) Molte delle cose che abbiamo detto nel paragrafo Alcune molecole biatomi che eteronucleari trattate con il metodo O.M. sono ancora perfettamente applicabili. Si vada a rileggere l’introduzione al paragrafo.
(5) Ricordiamo che gli orbitali atomici solo quando hanno lo stesso segno si possono comporre per dare orbitali molecolari leganti (allo scopo si rivedano le figure 13 e 14).
(6) Anche se nel carbonio non figura il 2pz (poiché privo di elettroni), esso è sempre presente.
(7) Con i subindici a e b abbiamo indicato i due diversi atomi di ossigeno.
(8) Come hanno mostrato Hückel (1937) e Coulson (1941)
Categorie:Atomi e Molecole

Rispondi