1 – Molecole poliatomiche trattate con il metodo O.M.: orbitali molecolari localizzati e delocalizzati
Fino ad ora, in tutte le molecole delle quali ci siamo occupati, abbiamo fatto esplicito riferimento ad un tipo di legame che prevedeva la localizzazione di una coppia di elettroni, e quindi dell’orbitale molecolare che li rappresentava, in una zona bea precisa di spazio, compresa al massimo tra due nuclei. Più in generale, elettroni che siano confinati in un orbitale atomico di un atomo o nella regione di legame tra due atomi in un orbitale molecolare (soprattutto negli orbitali di tipo σ più volte incontrati), originano orbitali localizzati.
La teoria L.V. usa esclusivamente di orbitali localizzati oltre al fondamentale concetto di risonanza(1) al quale, come abbiamo visto, corrisponde una quantità di energia di legame dipendente dal numero di strutture tra le quali la molecola risuona.
Anche la teoria O.M. si serve di orbitali molecolari localizzati, ed anzi quando ciò è possibile è preferibile, ma spesso si ha a che fare con sistemi per i quali è difficile mettere insieme una coppia di elettroni in un orbitale molecolare unico e quindi localizzato. In questo caso si deve introdurre il concetto di orbitali (molecolari) delocalizzati, l’analogo nella teoria O.M. di ciò che rappresenta la risonanza nella teoria L.V. (come dovrebbe essere ormai chiaro la teoria O.M. non si serve della risonanza). Quando un orbitale molecolare non è più un semplice orbitale molecolare, così come lo abbiamo descritto nelle pagine precedenti, ma è il risultato di una mescolanza tra orbitali molecolari ed atomici(2) allora esso diventa un orbitale molecolare delocalizzato.
E’ chiaro da quanto appena eletto che su un orbitale delocalizzato vi possono essere più di due elettroni e, poiché questo orbitale è sempre il risultato del legame che si forma tra più di due atomi, esso sarà un orbitale che interessa l’intera molecola, estendendosi al di sopra (e al di sotto) di tutti i nuclei che ne fanno parte. Così intendendo le cose, non avrà più senso dire che una coppia di elettroni è localizzata tra due nuclei; poiché l’orbitale delocalizzato si estende su tutti i nuclei, gli elettroni che in esso si trovano possono essere situati in qualunque zona dell’orbitale (che avrà simmetria di tipo p, con i nuclei giacenti nel piano nodale), conseguentemente in qualsiasi zona della molecola (all’interno dell’orbitale) ed in definitiva sono liberi di muoversi in tutta la molecola (all’interno dell’orbitale). A questo libero movimento degli elettroni nell’orbitale vi è una sola limitazione ed è ancora dovuta al principio di Pauli: nelle vicinanze di un nucleo, su un dato livello energetico di quell’atomo, non possono esservi più di due elettroni e questo fatto limita le possibili posizioni degli elettroni nell’intera molecola.
E’ interessante osservare che alla delocalizzazione degli orbitali corrisponde un’energia di legame, detta energia di delocalizzazione, che, facendosi i conti, ha lo stesso valore dell’energia di risonanza della teoria L.V.
Alcune volte, anche nel caso di molecole poliatomiche, sarà possibile darne una descrizione con la teoria O.M. utilizzando orbitali molecolari localizzati; altre volte ciò è praticamente impossibile, come nel caso degli stati eccitati di molecole contenenti non più di un doppio legame ed in quello delle molecole aromatiche (ad esempio: il benzene C6H6) e coniugate (3) (ad esempio: il butadiene C4H6), e bisognerà ricorrere agli O.M. delocalizzati.
Di seguito vedremo degli esempi di un caso e dell’altro, trattando di varie molecole poliatomiche (già trattate con il metodo L.V.: proprio per porre in evidenza le differenze di trattazione tra i due metodi), e dagli esempi risulterà anche più chiaro quanto abbiamo fin qui detto a proposito di O.M. localizzati e delocalizzati.
Naturalmente si opererà anche qui in approssimazione L.C.A.O.
BeH2 – molecola di idruro di berillio. (O .M. delocalizzati)
Questa, semplice molecola poliatomica, che già abbiamo visto essere lineare, ci offrirà lo spunto per vedere come il metodo O.M. vada applicato a molecole con più di due atomi.(4)
Cominciamo con il ricordare la configurazione elettronica del berillio:
Be[(1s2)(2s2)] => (eccitando) Be[(1s2)(2s)(2px)];
gli orbitali di valenza del berillio sono quindi il 2s ed il 2px, mentre l’idrogeno ha l’orbitale di valenza 1s. Si tratterà quindi di combinare, in orbitali molecolari, gli orbitali atomici 2s e 2px del berillio con i due orbitali atomici 1s dei due idrogeni (che per comodità indichiamo con Ha ed Hb). Si devono cioè trovare delle funzioni d’onda molecolari che siano opportune combinazioni lineari delle funzioni d’onda atomiche e nel far ciò è molto importante tener conto del segno che le funzioni d’onda atomiche (o gli orbitali atomici) hanno. Con un disegno possiamo capire meglio: serviamoci quindi di figura 42. La figura 42a mostra, la sovrapposizione dell’orbitale 2s del berillio con gli orbitali 1s
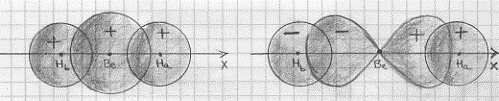
Figura 42
degli idrogeni. Come si può vedere si hanno sempre gli stessi segni per gli orbitali atomici e, conseguentemente, la frazione d’onda molecolare (legante) che rappresenterà questa situazione sarà, a meno di fattori moltiplicativi costanti detti di normalizzazione, la semplice somma delle singole funzioni d’onda atomiche:
(1) ψ (σ2s)= ψ Hb (1s) + ψBe (2s) + ψ Ha (1s)
La figura 42b mostra la sovrapposizione dell’orbitale 2px del berillio con gli orbitali 1s degli idrogeni. Qui ci troviamo di fronte ad una situazione differente da quella appena vista. Ora il 2px del berillio ha un lobo positivo ed uno negativo mentre gli 1s degli idrogeni sono sempre positivi. Per poter fare una combinazione lineare di questi orbitali atomici per ottenere un orbitale molecolare legante, occorrerà cambiare segno all’orbitale atomico 1s dell’idrogeno Hb(5) (a sinistra in figura 42b) e, per farlo, basta cambiare segno alla funzione d’onda che lo rappresenta. In questo caso, sempre a meno di fattori moltiplicativi costanti o di fattori di normalizzazione, la funzione d’onda molecolare (l’orbitale molecolare) legante sarà:
(2) ψ(σ2px) ~ ψ Ha (1s) + ψBe (2px) – ψ Hb (1s)
Con un analogo ragionamento siamo in grado di costruirci gli orbitali molecolari antileganti σ*. Si tratta di fare una sovrapposizione come quella mostrata in figura 42, solo che ora occorrerà cambiare i segni degli 1s degli idrogeni rispetto a quelli indicati nella figura 42. Si ha allora:
(3) ψ (σ*2s) ~ – ψ Hb (1s) + ψBe (2s) – ψ Ha (1s)
(4) ψ (σ*2px) ~ – ψ Ha (1s) + ψBe (2px) + ψ Hb (1s)
Ricordando ora quanto abbiamo detto a proposito delle combinazioni lineari è più corretto scrivere le (l), (2) ,(3) e (4) nel modo seguente:
ψ (σ2s) = ψ Be (2s) + k1[ψHa (1s) + ψ Hb (1s)]
ψ (σ2px) ~ ψ Be (2px) + k2[ψHa (1s) – ψ Hb (1s)]
ψ (σ*2s) ~ ψ Be (2s) – k3[ψHa (1s) + ψ Hb (1s)]
ψ (σ*2px) ~ ψ Be (2px) – k4[ ψBe (1s) – ψ Hb (1s)]
dove k1 , k2 , k3 e k4 sono, al solito, delle costasti che ci danno indicazioni sul peso relativo degli orbitali atomici di valenza del berillio e dell’ idrogeno nella formazione della molecola; esse vanno determinate a partire dalla condizione che l’energia deve risultare minima, affinché il legame sia stabile. Facendo i conti, che sono al di fuori della nostra portata, si trova che le superfici limite degli orbitali molecolari σ e σ*, per la molecola di BeH2 , sono quelli riportati in figura 43. Inoltre, poiché risulta che gli orbitali molecolari più stabili sono quelli leganti, i 4 elettroni di valenza della molecola di BeH2 si sistemeranno in essi e, in definitiva, la configurazione elettronica della molecola sarà:
BeH2[k(s2s)2(s2px)2]
e, come si vede, le due coppie di elettroni che formano il legame sono delocalizzati sui tre atomi che formano la molecola (non c’è modo di localizzare le coppie di elettroni).
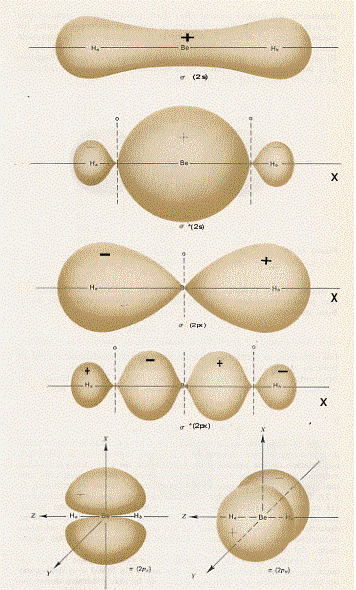
Figura 43
CO2 – Molecola di anidride carbonica (O.M. delocalizzati)
Ricordiamo le configurazioni elettroniche del carbonio e dell’ossigeno:
C[(1s2)(2s2)(2px)(2py)] ; O[(1s2)(2s2)(2px)(2py)(2pz2)]
Gli orbitali di valenza del carbonio sono il 2s , il 2px ed il 2py ; quelli dell’ossigeno sono il 2px , il 2py ed il 2pz (avendo trascurato per semplicità il 2s2 ). Come si ricorderà anche questa è una molecola lineare.
La sovrapposizione degli orbitali 2s e 2px del carbonio con i due orbitali 2px dei due ossigeni ci forniranno i legami di tipo σ e σ*; la sovrapposizione degli orbitali 2py e 2pz(6) dei tre atomi ci fornirà i legami di tipo π e π* . Le combinazioni lineari degli orbitali atomici che ci forniscono gli orbitali molecolari saranno allora:(7)
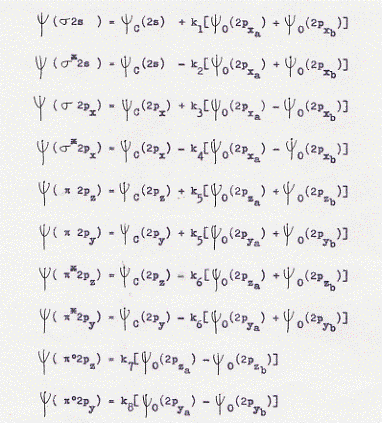
Le combinazioni lineari cosi ottenute sono in numero di 10. Per le prime 8 non c’è altro da fare che la figura che ne rappresenta le superfici limite (figura 44), poiché la formazione degli orbitali molecolari avviene allo stesso modo che abbiamo visto nel caso di BeH2, con la sola differenza che ora sono i 2px degli ossigeni anziché gli 1s degli idrogeni a formare orbitali molecolari σ e σ*. Le ultime due combinazioni lineari abbisognano invece di una spiegazione.
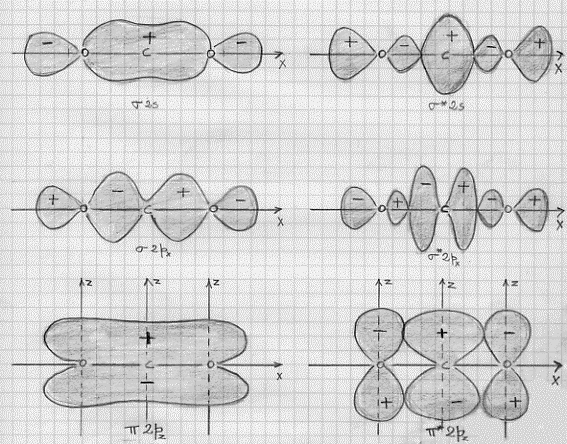
Figura 44 – Non sono stati disegnati i π2py e π*2py perché equivalenti, rispettivamente, ai π2pz e π*2pz .
Discuteremo solo la prima delle due, poiché per l’altra vale un discorso esattamente equivalente, servendoci della figura 45a nella quale sono rappresentati gli orbitali atomici 2pz di ciascuno dei tre atomi che vanno a formare la molecola. La figura corrisponde alla combinazione lineare:
ψ (π2pz)= ψ C (2pz) + k7[ ψO (2pza) – ψ O (2pzb)]
e, come si può vedere dalla figura, tra i vari 2pz non vi è una sovrapposizione netta a causa del segno di 2pzb che,
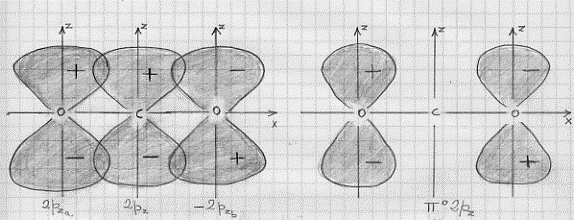
Figura 45
essendo negativo, non permette che si abbia una ψ interamente positiva al di sopra dell’asse x ed interamente negativa al di sotto del medesimo asse. Di conseguenza la cosa va come se non vi fosse alcuna sovrapposizione con l’orbitale 2pz del carbonio e si dovessero considerare solo i 2pz degli ossigeni. Rimane allora solo la combinazione 2pza – 2pzb che equivale a porre ψ C (2pz) = 0 nella relazione precedentemente scritta; e questa combinazione è anch’essa un orbitale molecolare costituito da due orbitali atomici che praticamente non interagiscono tra di loro per la distanza a cui si trovano. E se non c’è interazione, non c’è cambio nel valore dell’energia (si riveda la figura 12) che resta quello che compete agli orbitali atomici dei due ossigeni. In questo modo l’orbitale molecolare risultante non è né legante né antilegante: esso è detto non legante ed indicato ,con il simbolo π°2pz (si veda figura 45b).
In definitiva, la configurazione elettronica di CO2 è data da:
CO2[KK(2sa2)(2sb2)(σ2s)2(σ2px)2(π2py)2(π2pz)2(π°2py)2(π°2pz)2],
ed anche qui gli orbitali molecolari risultano completamente delocalizzati. Si deve notare che l’energia di π2py è la stessa di π2pz, che l’energia di π°2py è la stessa di π°2pz che come al solito gli altri valori dell’energia sono scritti in successione crescente e che non vi sono elettroni in orbitali antileganti.
BF3 – molecola di trifluoruro di boro (O.M. delocalizzati)
Iniziamo con il ricordare che questa molecola ha una struttura piana trigonale e che le configurazioni elettroniche del boro e del fluoro sono:
B[(1s2)(2s2)(2px)] ; F[(1s2)(2s2)(2px)(2py2)(2pz2)]
saranno quindi gli orbitali atomici 2s e 2px del boro a legarsi in orbitali molecolari con gli orbitali atomici 2s e 2px del fluoro. Ricordiamo anche che, se pure non compaiono nella configurazione elettronica del boro, occorrerà prendere in considerazione anche i suoi orbitali atomici 2py e 2pz che, come sappiamo, sono vuoti. Poiché poi abbiamo a che fare con tre atomi di fluoro, ci serviremo dei subindici a, b, c per indicarli. Infine dobbiamo osservare che le coordinate x, y, z per gli orbitali atomici del boro non coincidono con quelle del fluoro (proprio perché la molecola è trigonale piana).
Fatte queste premesse, iniziamo con il disegnarci queste sovrapposizioni e quindi a costruirci gli orbitali molecolari σ, dovuti alla sovrapposizione degli orbitali atomici 2s, 2py e 2pz del boro con gli orbitali 2pxa , 2pxb , 2pxc dei fluori, ed infine i π, dovuti alla sovrapposizione del 2pz del boro e dei 2py dei fluori. Nella figura 46 sono riportate:
in (a) le sovrapposizioni del 2s del boro con i 2px dei fluori;
in (b) quelle del 2py del boro con i 2px dei fluori;
in (c) quelle del 2px del boro con i 2px dei fluori;
in (d) quelle del 2pz del boro con i 2py dei fluori;
in (e) ed in (f) due possibili orbitali non leganti che anche in questo caso si producono e che discuteremo tra poco.
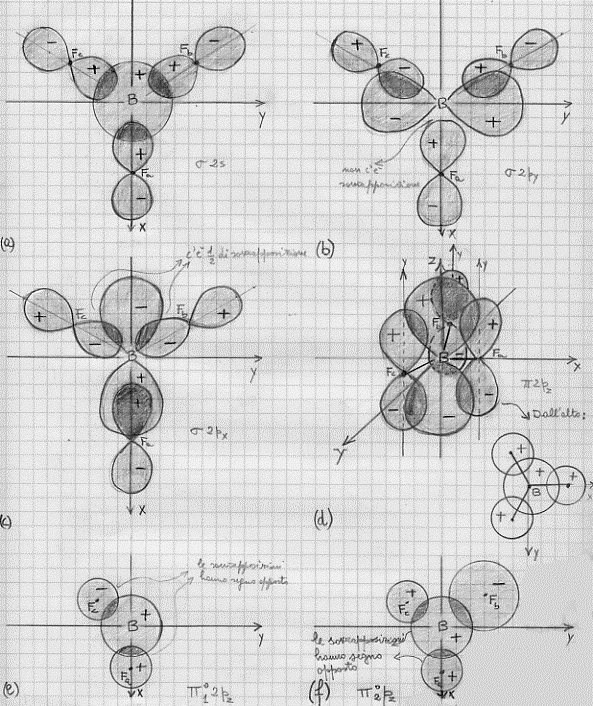
Figura 46
Le funzioni d’onda molecolari, combinazioni lineari delle funzioni d’onda atomiche, che ci rappresentano ordinatamente le situazioni di figura 46, sono:
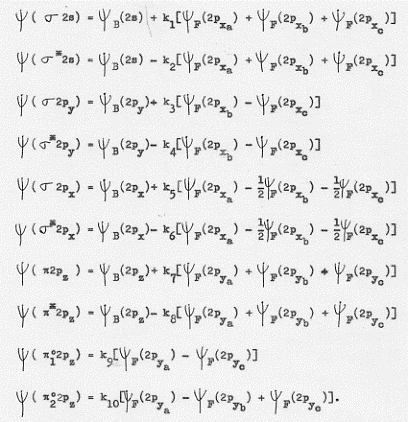
(Qui, come anche nella fig. 46, compare uno ‘strano’ orbitale molecolare σ2py. Ebbene esso non ha nulla a che vedere con gli orbitali molecolari definiti in figura 14, tanto più che questo tipo di orbitale non esisteva proprio – gli orbitali di tipo σ potevano essere realizzati solo dalla sovrapposizione di orbitali atomici di tipo s o px -. Gli orbitali molecolari dei quali discutiamo ora (caso di molecole poliatomiche) sono originati da diverse sovrapposizioni tra orbitali atomici di tipo differente ed essi saranno σ o π, a seconda della forma risultante).
Non resta ora che dare la configurazione elettronica di BF3:
BF3[KK(2sa2)(2sb2)(σ2s)2(σ2py)2(σ2px)2(π2pz)2(π°12pz)2(π°22pz)2(2pza2)(2pzb2)(2pzc2)],
come si vede, vi sono 6 elettroni in orbitali leganti σ e ciò vuol dire che si hanno 3 legami σ; vi sono inoltre due elettroni in un orbitale legante π che formano un legame π che risulta completamente delocalizzato (i due elettroni che formano il legame π appartengono all’intera molecola). Si può rappresentare questo legame π delocalizzato come in figura 47.
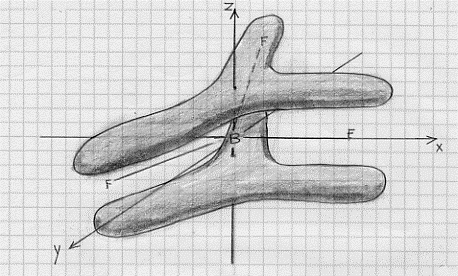
Figura 47
H2O – molecola d’acqua (O.M. delocalizzati)
Senza entrare in troppi dettagli, si tratterà di costruire delle combinazioni lineari degli orbitali atomici 2s, 2px , 2py dell’ossigeno con gli 1s degli idrogeni. Nel far questo dovremo tener conto che sperimentalmente l’angolo di legame risulta di circa 105°.
Le possibili sovrapposizioni (combinazioni lineari) leganti sono mostrate in figura 48. Si deve però tener conto che le ultime due sovrapposizioni, σ2px e σ2s (figura 48b e 48c), avendo la stessa simmetria, si combinano tra di loro in una specie di ibridizzazione per formare un orbitale molecolare fortemente legante (σ2s) ed uno con carattere parzialmente non legante, che chiameremo σ x. Vi sono poi i due elettroni dell’ossigeno che si trovano sull’orbitale atomico 2pz; ma quest’ultimo non ha alcuna sovrapposizione con gli 1s degli idrogeni e pertanto si dovrà considerare un legame di tipo π non legante e cioè π°2pz . Infine si avranno gli orbitali molecolari non leganti (che si ottengono dalle
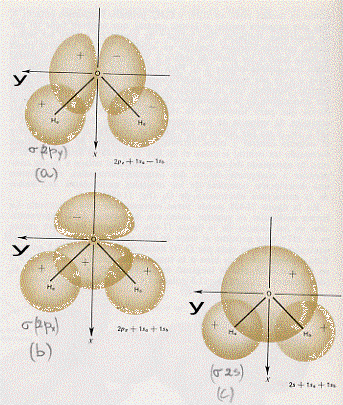
Figura 48
combinazioni mostrate in figura 48 quando si cambi segno agli ultimi tre termini) anche se su di essi non vi sono elettroni.
La configurazione elettronica di H2O sarà allora:
H2O[K(σ2s)2(σ2py)2(σx)2(π°2pz)2],
e, come si vede, si avranno due legami netti di tipo σ. Si deve però osservare che se si avessero solo questi due legami netti l’angolo di legame sarebbe di 90°. E’ proprio l’ulteriore parziale legame (il σx), come hanno mostrato recentemente Pitzer e Cusachs, che fa si che l’angolo di legame tenda ai quasi 105°. Da questa discussione risulta che 6 elettroni sono in orbitali molecolari delocalizzati nell’intera molecola mentre solo due (i π°2pz) risultano localizzati. Infine, questa descrizione è in accordo con le misure spettroscopiche poiché tutti e quattro gli orbitali risultanti hanno energie distinte (nessun orbitale è degenere – si veda figura 49), contrariamente a quanto accadeva nel caso della stessa molecola discussa con il metodo L.V. e mediante ibridizzazione sp (allora si avevano due orbitali molecolari leganti σ equivalenti in energia) e ciò vuol dire che è il metodo L.V. quello in accordo con l’esperienza spettroscopica.

Figura 49 – Si osservi che solo i primi 4 orbitali molecolari risultano pieni; quelli antileganti non contengono elettroni.
H2O – molecola d’acqua (O.M. localizzati)
Oltre alla trattazione della molecola d’acqua mediante gli orbitali molecolari delocalizzati, è possibile farne un’altra mediante l’uso degli orbitali molecolari localizzati. In questo caso occorrerà considerare la combinazione lineare degli orbitali atomici 1s degli idrogeni e degli orbitali 2px e 2py dell’ossigeno. Si può subito procedere ad una approssimazione (per altri versi molto ragionevole) che prevede separatamente la combinazione lineare dell’1s di un idrogeno con il 2px dell’ossigeno e dell’1s dell’altro idrogeno con il 2py dell’ossigeno.
Ricordando però che gli orbitali atomici 2px e 2py sono perpendicolari tra di loro ci si dovrebbe aspettare un angolo di legame di 90° (si veda figura 50 a), contrariamente al fatto, che già conosciamo e verificato sperimentalmente, che l’angolo di legame nell’acqua è di circa 105°. Per rendere conto di questa differenza (figura 50 b) si sono ricercate varie spiegazioni, tra le quali:
1) la repulsione elettrostatica tra gli atomi di idrogeno (elettrone-elettrone e protone-protone) che sono legati con l’ossigeno;
2) la repulsione tra i due legami O-H;
3) un particolare intervento delle coppie solitarie di elettroni (si riveda quanto abbiamo detto a proposito della molecola
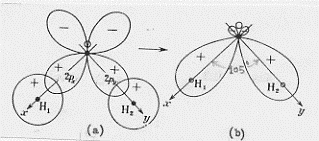
Figura 50
d’acqua quando la abbiamo trattata con il metodo L.V. mediante gli orbitali ibridi).
Una ultima cosa resta da dire a proposito della molecola d’acqua, almeno nell’ambito dei nostri scopi, ed è relativa al suo essere polare, al fatto cioè che ad essa si deve assegnare un momento di dipolo (md). Questo momento di dipolo è dovuto alla somma vettoriale del momento di dipolo originato dagli elettroni che formano il legame e del momento di dipolo originato dalle coppie di elettroni solitari. E mentre quest’ultimo è un unico momento di dipolo, l’altro è originato per somma vettoriale dei singoli momenti di dipolo di ciascun legame (si veda la figura 51).
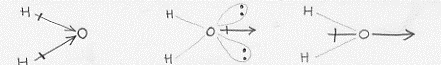
Dipoli di legame Dipolo di coppie solitarie Dipolo totale
Figura 51
C6H6 – molecola di benzene (O.M. delocalizzati )
La costruzione di questa molecola con la teoria degli orbitali molecolari si fa utilizzando in parte la teoria degli ibridi introdotta nel metodo L.V. ed in parte la teoria degli O.M. delocalizzati.
I 6 legami carbonio-carbonio ed i 6 legami carbonio-idrogeno si costruiscono esattamente nello stesso modo già discusso nel metodo L.V., mediante gli ibridi sp2 del carbonio e gli orbitali atomici 1s degli idrogeni. Questo procedimento ci fornisce i legami localizzati s che costituiscono lo scheletro della molecola (si riveda la figura 33). Restano ora da considerare i sei orbitali atomici 2pz dei sei atomi di carbonio (si riveda la figura 34) che ci forniranno i legami delocalizzati di tipo p. In effetti non vi è ragione di considerare una sovrapposizione piuttosto che un’altra di questi orbitali 2pz . Sembra lecito pensare ad una sovrapposizione cumulativa in modo da ottenere orbitali molecolari delocalizzati sull’intera molecola. E, dato che si hanno 6 orbitali atomici, si avranno 6 orbitali molecolari, 3 dei quali leganti e 3 antileganti. I 6 elettroni disponibili si troveranno sugli orbitali molecolari leganti che sono mostrati nella figura 52. Questi elettroni, liberi di muoversi nell’intera molecola, sono stati chiamati da Lennard-Jones (1937) elettroni mobili e Coulson ha proposto di considerarli come una piccola corrente elettrica che gira intorno all’anello e, poiché
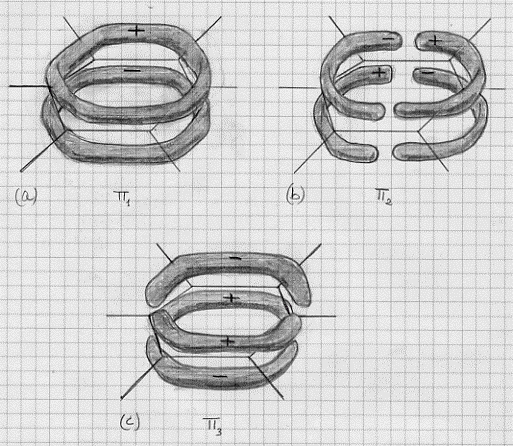
Figura 52
normalmente vi sono correnti uguali ruotanti in versi opposti, l’effetto totale sarà nullo. Ma si comincia qui a profilare un’analogia tra gli elettroni delocalizzati nel benzene e gli elettroni di conduzione nei metalli: in ambedue i casi una perturbazione elettrica provocata in una parte del sistema si propaga con facilità in altre parti.
Il fatto poi che gli elettroni abbiano una zona più ampia in cui muoversi fornirà un valore più basso per l’energia totale e quindi una più alta energia di legame rispetto al caso degli orbitali localizzati; si tratta proprio di quell’energia delocalizzazione (Coulson, 1947) che è l’analogo O.M. dell’energia di risonanza L.V.
– La grafite ed il diamante –
E’ ora interessante almeno un cenno a queste due forme di cristallizzazione del carbonio. E’ istruttivo andare a vedere, per differenza tra grafite e diamante, quale ruolo giocano gli orbitali molecolari delocalizzati.
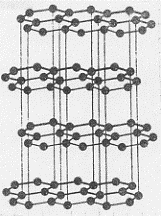
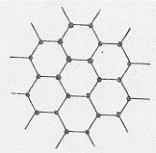
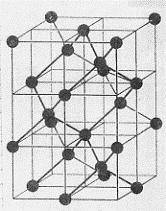
Iniziamo con la figura 53 che mostra la differenza tra le due strutture cristalline. Come si vede (figura 53b) la grafite, una delle forme solide nelle quali incontriamo il carbonio, ha una struttura cristallina a celle esagonali. Il
(a)
(b)
(c)
Figura 53
carbonio, tetravalente, si lega ai tre atomi di carbonio che gli giacciono vicini nello stesso piano mediante legami σ dovuti ad ibridizzazione trigonale sp . In questo modo si saturano 3 valenze. L’altro legame è dovuto agli orbitali atomici 2pz del carbonio (che risultano perpendicolari al piano di figura 53b). Questo legame è di tipo π ed è completamente delocalizzato su tutto il piano degli esagoni. Ogni stato di esagoni costituisce una lamina (una gigantesca molecola) che è legata alla lamina sovrapposta e sottoposta da deboli forze di Van der Waals. Si hanno in definitiva tante lamine sovrapposte (figura 53a): all’interno delle lamine vi sono forze intense contrariamente a ciò che avviene tra lamina e lamina. Quest’ultimo fatto spiega bene il potere lubrificante della grafite: sono le lamine che, scorrendo le une sulle altre, originano il fenomeno. Inoltre gli orbitali delocalizzati rendono conto del potere conduttore che questo materiale presenta.
Nel caso del diamante (figura 53c) quanto ora detto non si verifica poiché si ha cristallizzazione nella struttura tetraedrica a coppie di elettroni localizzati, dovuta ad ibridizzazione tetragonale sp3 che, come sappiamo, non lascia orbitali non mescolati. Questo tipo di cristallizazione, oltre a quante detto, rende conto della grande durezza del diamante e del suo potere isolante.
NOTE
(1) Approfitto dell’occasione per ricordare che se due (o più) sono le possibili strutture che noi utilizziamo per rappresentare una molecola, parlare di risonanza tra queste strutture significa solo dire che nessuna delle strutture rappresenta bene la situazione, la quale invece è meglio descritta dalla sovrapposizione (matematica) di tutte le possibili strutture (che sono solo dei modelli matematici senza alcuna realtà fisica).
(2) Si noti la differenza con gli ibridi: in quel caso si aveva mescolanza tra orbitali atomici e basta. Successi vamente questi nuovi orbitali atomici, gli ibridi, andavano a formare legami con altri orbitali atomici.
(3) Le molecole aromatiche sono quelle i cui atomi componenti sono disposti ad anello chiuso. Si hanno invece molecole coniugate quando in esse sono presenti due doppi legami separati da un legame semplice; ad esempio il butadiene ha la seguente formula di struttura: CH2=CH—CH=CH2 .
(4) Molte delle cose che abbiamo detto nel paragrafo Alcune molecole biatomi che eteronucleari trattate con il metodo O.M. sono ancora perfettamente applicabili. Si vada a rileggere l’introduzione al paragrafo.
(5) Ricordiamo che gli orbitali atomici solo quando hanno lo stesso segno si possono comporre per dare orbitali molecolari leganti (allo scopo si rivedano le figure 13 e 14).
(6) Anche se nel carbonio non figura il 2pz (poiché privo di elettroni), esso è sempre presente.
(7) Con i subindici a e b abbiamo indicato i due diversi atomi di ossigeno.
(8) Come hanno mostrato Hückel (1937) e Coulson (1941).
Categorie:Senza categoria

Rispondi