1 – Le forze di Van der Waals. Le forze di dispersione di London
Bisognava proprio aspettare il 1930. Bisognava aspettare che la meccanica quantistica avesse raggiunto una completa formulazione ad opera di Schrödinger e di Heisenberg nel 1926, affinché si potesse giungere ad una più soddisfacente comprensione delle forze intermolecolari.
London comincia ad occuparsi di problemi di meccanica quantistica nel 1926 con un articolo riguardante la densità di carica degli elet troni negli atomi e nelle molecole. In precedenza (1925) si era occupato insieme a H. Hönl del problema dell’intensità delle bande degli spettri molecolari. Nel 1927, insieme a W.Heitler, pubblica un fondamentale lavoro relativo alla trattazione quantistica del legame covalente (introdotto empiricamente da Lewis nel 1916. Su questa trattazione, nota come teoria del legame di valenza torneremo diffusamente più oltre). Nel 1928 il fisico tedesco torna ripetutamente sull’argomento (legame covalente) ampliandolo e perfezionandolo. Nel 1930 pubblica un primo articolo insieme a R.Eisenschitz in cui mette in relazione le forze di covalenza con le forze di Van der Waals. Finalmente, ancora nel 1930, pubblica due articoli estremamente importanti in cui studia diffusamente (da un punto di vista teorico e sperimentale) le forze di Van der Waals dandone una spiegazione quanto-meccanica in grado di eliminare i dubbi lasciati dalle trattazioni di Keesom e Debye.
Seguiamo il lavoro di London partendo dalla critica da lui fatta ai risultati di Keesom e Debye.
La più ovvia obiezione che si può fare agli studi di Keesom (forze di orientamento) e Debye (forze di induzione) sulle forze molecolari è il non rendere conto del fatto che queste forze (forze di Van der Waals) si possono considerare come la causa comune di molti fenomeni diversi tra loro: identità delle forze molecolari negli stati liquido e gassoso (ricordo che il titolo del lavoro di Van der Waal s in cui si introduce la sua equazione e le sue forze è: Sulla continuità dello stato liquido e gassoso); capillarità e adsorbimento (mentre con assorbimento si intende l’incamerare, ad esempio, una molecola all’interno di un certo materiale, con adsorbimento si intende che, ad esempio, una molecola rimane trattenuta dalla superficie di quel certo materiale); calori di sublimazione di reticoli molecolari; certi effetti di allargamento delle righe degli spettri molecolari; etc… Non si riesce a capire come, per esempio, le stesse forze che agiscono nei liquidi e nei solidi fra molte molecole vicine debbono allo stesso modo agire fra le coppie occasionali di molecole in un gas.
In realtà i modelli di Keesom e Debye non sono in grado di spiegare una generale additiva coesione (si può dimostrare che i due tipi di forze non sono additivi) come quella che si manifesta in un solido o in un liquido.
Supponiamo di avere due molecole A e B (con dipoli permanenti o indotti). Supponiamo poi che queste due molecole siano orientate in modo tale da essere attratte da una terza molecola C. Le molecole A e B dovranno allora essere orientare tra di loro in modo da respingersi, infatti, se 1’orientamento di C è come in figura (a), le orientazioni di
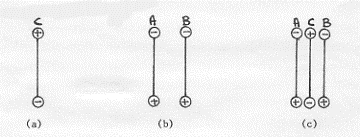
A e B (per essere attratte da C) dovranno essere come in figura (b) e quindi, mettendo insieme le tre molecole come in figura (c), si dovrà avere un effetto repulsivo tra A e B.
Allora, se le forze in gioco tra le molecole sono dovute alla polarizzazione, quando molte molecole da posizioni differenti sovrappongono i loro campi polarizzanti, il campo polarizzante totale dovrà usualmente risultare molto più piccolo di quello che si avrebbe se, ad esempio, le molecole fossero due sole. Ci si aspetterebbe quindi che in un solido o in un liquido le forze molecolari, originate da dipoli indotti o permanenti (ed anche quadrupoli od ottupoli), dovrebbero risultare diminuite di molto se non addirittura annullate per ragioni di simmetria.
La situazione risulta ancora peggiore se ci riferiamo ai gas rari i quali non hanno né dipoli permanenti, né quadrupoli, né ottupoli ma risultano avere una simmetria perfettamente sferica. I gas rari, come del resto abbiamo già detto, non presentano né interazioni di Keesom, né interazioni di Debye.
Per quanto riguarda poi le molecole di idrogeno (H2 ) e di azoto (N2 ), esse, pur essendo dotate di momento di quadrupolo, manifestano delle forze attrattive tra gli atomi costituenti che sono circa 100 volte maggiori di quelle che si possono calcolare considerando i quadrupoli ad esse associati.
Come si vede, quindi, le cose non vanno: occorre introdurre la meccanica quantistica per rimuovere le gravi obiezioni che sono state mosse.
Abbiamo già detto che nel 1900 Max Planck introdusse il quanto di energia (E = hn) , come artificio di calcolo, studiando l’emissione di radiazione da parte di un corpo nero (nel sito vi è una discussione molto ampia su queste vicende).
Secondo i conti fatti da Planck l’energia che compete ad un oscillatore armonico (una carica od un atomo oscillante armonicamente ed in prima approssimazione il moto degli atomi nelle molecole)(1) è quantizzata, può cioè assumere solo una serie di valori discreti (multipli di hn) e non tutti i possibili valori del continuo (oscillatore armonico quantizzato).
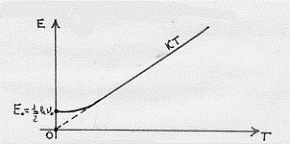
Sviluppando i conti si trova che un oscillatore armonico quantizzato, anche allo zero assoluto (T = 0 °K = -273 °C), possiede una energia diversa da zero (e questo fatto è completamente contrastante con quanto ricavabile dalla meccanica classica: allo zero assoluto l’energia è nulla risultando,classicamente: E proporzionale a KT, essendo K la costante di Boltzmann che abbiamo già incontrato). L’energia E che un oscillatore armonico (unidimensionale) ha allo zero assoluto vale esattamente mezzo quanto di Planck:
E0 = ½ hn0
ed a questa energia si dà il nome di energia di punto zero. Vediamo con un grafico (energia E dell’oscillatore in funzione della temperatura T) il confronto a basse temperature tra i risultati classici (linea tratteggiata) e quantistici (linea continua), con l’osservazione che a temperature T relativamente alte i risultati classici e quantistici coincidono::
Quanto abbiamo ora detto (ricavato appunto, senza giustificazione teorica, da Planck nel 1900) è in completo accordo con il principio di indeterminazione di Heiseinberg del 1927. Vediamo come e perché.
Abbiamo detto che il minimo di energia che ha un oscillatore armonico unidimensionale (pensiamo alle oscillazioni degli elettroni degli atomi costituenti una molecola vale E0 = ½ hn0 (2) (e questa è l’energia che corrisponde allo stato fondamentale, cioè non eccitato, degli elettroni negli atomi che costituiscono la molecola). Questo minimo di energia è chiamato energia di punto zero dell’oscillatore. Il minimo di energia calcolato secondo la meccanica classica corrisponde al punto 0 della curva di energia potenziale dell’ultima figura di Nota 2. Comunque, in corrispondenza di questo punto (classico), si ha x = 0 e v = 0 (per la posizione e la velocità dell’oscillatore in considerazione). Poiché non ci dovrebbe essere alcuna oscillazione in questa posizione, noi ci troveremo nelle condizioni di conoscere contemporaneamente e con assoluta precisione la posizione e la velocità degli elettroni oscillanti: questo fatto è però in contraddizione con il principio di indeterminazione di Heisenberg. Allora il primo livello energetico (dello stato fondamentale dell’oscillatore) dovrà essere il più basso livello energetico compatibile con in principio di indeterminazione (livello E della figura citata).(3)
Dopo questa necessaria digressione sull’energia di punto zero, torniamo alle forze intermolecolari.
Consideriamo, con London, due sistemi a simmetria sferica, ciascuno con una certa polarizzabilità a, i quali siano degli oscillatori armonici tridimensionali (con tre gradi di libertà) con nessun momento di dipolo o quadrupolo o ottupolo nelle loro posizioni di riposo (ad esempio: due atomi di un gas raro). Classicamente i due sistemi nelle loro posizioni di equilibrio non dovrebbero interagire e quando li avviciniamo dovrebbero rimanere nelle loro posizioni di riposo senza indurre nessun momento (di dipolo o altro) l’uno sull’altro.
Quantisticamente abbiamo visto che una particella è dotata sempre, nel suo stato di riposo, di una energia di punto zero (è dotata cioè di movimento). Facendo i conti quanto-meccanici per questo sistema di due oscillatori si trova che lo stato fondamentale E0 dell’energia del sistema sarà dato da:
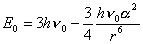
[si noti che se si annullasse l’energia di punto zero, si annullerebbe l’intera espressione]. Vediamo di descrivere questa relazione: innanzitutto il termine 3hn0 è l’energia di punto zero del sistema dei due oscillatori, ogni oscillatore ha tre gradi di libertà e quindi, essendo 6 i gradi complessivi di libertà per i due oscillatori, si ha che l’energia di punto zero vale 6.(½ hn0 ) = 3hn0 ; il secondo termine:
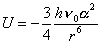
(dove r è la distanza tra i due oscillatori ed a la loro polarizzabilità)(4) dipende dalla distanza r tra i due oscillatori e può essere considerato come una energia di interazione tra gli oscillatori che, essendo negativa, caratterizza una forza attrattiva.
Possiamo subito immaginare allora che questo tipo di forza,(5) che non è condizionata dall’esistenza di nessun tipo di dipolo (o quadrupolo o ottupolo) permanente, sarà responsabile dell’attrazione di Van der Waals dei gas rari ed anche delle semplici molecole H2 , N2 , etc. Queste forze sono chiamate forze di dispersione(6). Benché non sia possibile, come del resto si può intuire, descrivere questo meccanismo di interazione in termini della nostra usuale meccanica classica, possiamo illustrarlo servendoci ancora di un linguaggio semiclassico.
Se si fosse in grado di fare una fotografia di una molecola ad un dato istante, si troverebbero varie configurazioni dei nuclei e degli elettroni (degli atomi costituenti)(7) tali da fornire, in generale, momenti di dipolo. In una molecola costituita da atomi di gas rari (molecola a simmetria sferica), la media su molte foto fatte alle configurazioni assunte dai nuclei e dagli elettroni (degli atomi costituenti) non dovrebbe dare nessuna preferenza ad un particolare dipolo in una privilegiata direzione (non dovrebbe esserci cioè nessun momento di dipolo privilegiato).
Questi dipoli (dipoli istantanei), che variano molto rapidamente nella molecola e che esistono e sono rappresentati grazie all’energia di movimento di punto zero della molecola stessa, producono un campo elettrico che agisce sulla polarizzabilità dell’altra molecola generandovi dei dipoli indotti che sono in fase ed interagenti con i dipoli istantanei che li hanno generati. L’energia di punto zero è, per così dire, accompagnata da un campo elettrico variabile alternativamente in sincronismo con essa; il campo elettrico variabile non emette però alcuna energia poiché, come abbiamo già detto, l’energia di punto zero non può essere dissipata in alcun modo essendo solo una frazione (½ hn0) del quanto di radiazione (hn0) di Planck (che è la minima quantità di energia che può essere emessa o assorbita) .
Vediamo ciò in un modo più descrittivo aiutandoci con delle figure.
Consideriamo due atomi di elio (che è un gas nobile di simbolo chimico He) .
L’atomo di elio ha due elettroni (situati nello stato fondamentale nell’orbitale 1 s a simmetria sferica) ciascuno dei quali lo indicheremo con – ed inoltre nel suo nucleo (tra l’altro) vi sono due protoni che indicheremo con + 2 . Questo atomo ha una simmetria sferica come mostrato in figura:

In ogni istante, comunque, gli elettroni occuperanno una determinata posizione dello spazio. La figura rappresenta le posizioni istantanee degli elettroni in molti differenti istanti di tempo (o, che è lo stesso, in molti differenti atomi allo stesso istante di tempo). Tra le diverse possibili con figurazioni degli elettroni rispetto al nucleo prendiamone in considerazione due, in due diversi istanti, come quelle riportate nella figure (a) e (b):
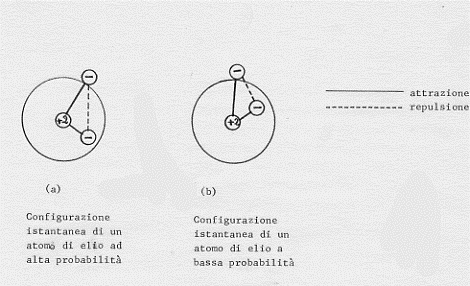
I due elettroni, nello stato fondamentale, stanno nell’orbitale 1 s che, appunto, è a simmetria sferica.
La figura (a) riporta una configurazione ad alta probabilità in cui gli elettroni si trovano in posizioni opposte rispetto al nucleo.
La figura (b) riporta una configurazione a bassa probabilità in cui gli elettroni si trovano ambedue da una stessa parte del nucleo.
Questa tendenza degli elettroni a sistemarsi in bande opposte rispetto al nucleo è chiamata correlazione elettronica ed è il risultato della repulsione tra cariche uguali. Questa repulsione rende quindi meno probabile la configurazione, a cui compete maggiore energia, di figura (b) rispetto a quella di figura (a), a cui compete minore energia (fatto in accordo con la tendenza di natura).
Naturalmente il fenomeno della correlazione elettronica si ha anche quando, anziché considerare un atomo isolato, si considera un aggregato di atomi. Il risultato di questa correlazione è stato riportato schematicamente nelle figure (a) e (b) seguenti, in cui è riportato un caso in cui c’è attrazione tra due atomi di elio ed un caso in cui c’è repulsione:
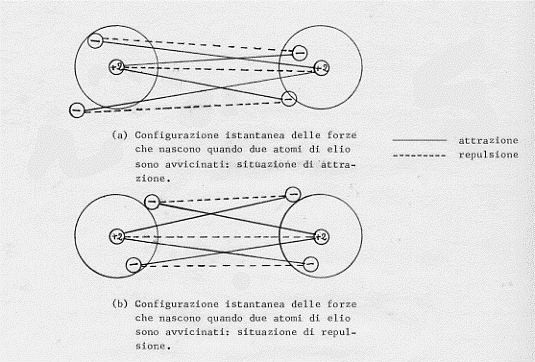
Ambedue i disegni mostrano le probabili disposizioni degli elettroni nei singoli atomi ma in situazione di differente orientazione di un atomo rispetto all’altro. Nella figura (a) le attrazioni fra elettroni di un atomo e nucleo dall’altro dominano sulle repulsioni elettroni-elettroni e nucleo-nucleo: a queste configurazioni corrisponde un abbassamento di energia. Nella figura (b) si ha una situazione opposta a cui corrisponde un innalzamento di energia.
La cosa importante da notare a questo punto è che gli elettroni dei due atomi correlano i loro movimenti in modo tale da rendere più probabile la configurazione di figura (a) ( a cui corrisponde una energia più bassa); e quindi si hanno sempre configurazioni il cui risultato è una attrazione.
Cerchiamo di capire ancora meglio.
Le configurazioni di figure (a) e (b) (configurazioni istantanee), possono essere considerate come semplici dipoli elettrici (istantanei) . Alla luce di questo fatto analizziamo di nuovo le interazioni di figure (b) e (c) con i seguenti disegni:
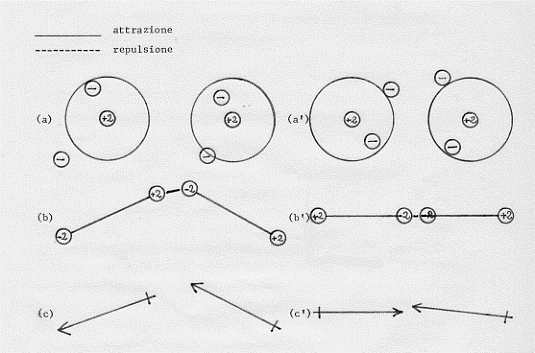
La figura mostra come si semplificano le cose considerando gli atomi come dipoli istantanei. Nella figura si passa da una configurazione atomica istantanea ad una configurazione dipolare istantanea.
In (a), (b) e (c) c’è, al solito, attrazione; mentre in (a’), (b’) e (c’) c’è repulsione [e noi sappiamo che è preferita, per ragioni energetiche, la configurazione (a), (b) e (c)].
In definitiva le cose stanno nel modo seguente: un atomo con una distribuzione di carica a simmetria sferica può essere considerato come un dipolo istantaneo; l’altro atomo che si avvicina al primo, poiché è dotato di una certa polarizzabilità, acquista un dipolo indotto che è in fase con quello inducente (in modo da originare sempre una attrazione).
Il motivo per cui le forze che si originano dalla vicinanza di due atomi (ad esempio) di elio sono sempre attrattive può essere riassunto nel modo seguente: consideriamo due atomi di elio in posizioni tanto ravvicinate da interagire; in questi atomi le posizioni degli elettroni non saranno più indipendenti ma, a causa della loro repulsione mutua, si avrà una maggiore probabilità di trovarli dallo stesso lato di ciascun nucleo (figura (b) seguente) o uno da un lato ed uno da
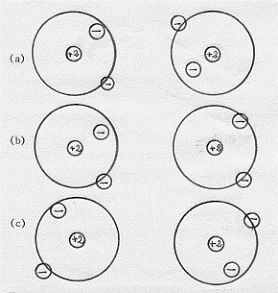
un altro (figura (c)) di quella di trovarli nella regione compresa tra i due nuclei (figura (a)); ragionando poi come due figure fa, si vede subito che alla configurazione di figura (a) corrisponde repulsione tra gli atomi, mentre a quelle di figure (b) e (c) corrisponde attrazione.
A questo punto è rimasta in sospeso la questione dell’additività delle forze di London. Cerchiamo di descrivere questo fatto.
Abbiamo già detto che una delle obiezioni che si possono muovere alle teorie di Keesom e Debye è il fatto che le forze di interazione molecolare, che si ricavano dallo sviluppo delle loro ipotesi, non risulta no additive.
Questo fatto non si verifica per le forze di London, infatti si può dimostrare che la relazione (che abbiamo già incontrato e che ci dà l’energia di attrazione tra gli atomi o le molecole):
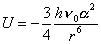
ha la caratteristica dell’additività; questo fatto significa che se tre molecole agiscono simultaneamente l’una sull’altra, i tre potenziali di interazione U fra le tre coppie possibili di molecole (se le molecole sono A, B e C, le tre possibili coppie sono AB, AC e BC) debbono essere semplicemente sommati, e che ogni influenza di una terza molecola sull’interazione fra le prime due è solo un piccolo effetto di perturbazione di un ordine di grandezza più piccolo di quello dell’interazione stessa.
Queste forze di interazione possono quindi essere semplicemente sovrapposte in accordo con la regola del parallelogramma delle forze ed esse sono, di conseguenza, in grado di rappresentare il fenomeno di generale coesione che si manifesta ad esempio tra liquidi e solidi.
Se molte molecole interagiscono simultaneamente tra di loro, si deve immaginare che ciascuna molecola induce su ciascuna delle altre un set di dipoli(8) che sono in costante relazione di fase con il corrispondente originale dipolo inducente. Ogni molecola è così sede si moltissimi set di dipoli indotti, sovrapposti in modo incoerente e originati dalle differenti molecole agenti. Ciascuno di questi dipoli indotti ha sempre un’orientazione tale da essere attratto dai dipoli corrispondenti che lo hanno generato. Si può così immaginare che l’interazione simultanea di molte molecole può semplicemente essere costruita successivamente come una sovrapposizione additiva di singole forze fra coppie di molecole.
Tutte le cose a questo punto sembrano tornare anche se c’è da osservare che l’ultima formula che abbiamo dato per l’energia di interazione U fra coppie di atomi o molecole è ancora lontana dal rappresentare le forze molecolari per tutte le distanze tra gli atomi o le molecole che interagiscono e può essere considerata solo come una prima approssimazione. Volendo infatti fare un conto più preciso occorrerebbe considerare anche i quadrupoli e gli ottupoli che originano interazioni proporzionali a 1/r8 ed 1/r10, etc., ma per i nostri scopi è più che sufficiente quanto abbiamo detto fino ad ora.
Una cosa è invece importante aggiungere relativamente all’interazione a piccole distanze r tra gli atomi o molecole.
Quando infatti due oscillatori interagenti si trovano ad una piccolissima distanza si verifica una penetrazione, l’una nell’altra.delle nuvole elettroniche di carica che circondano i nuclei.
In seguito a ciò i nuclei non risultano più completamente schermati dalle loro orbite elettroniche, si respingono l’un l’altro e danno origine ad una repulsione coulombiana.
Oltre a questo effetto repulsivo occorre considerarne un altro simultaneo: quello originato dal principio di esclusione di Pauli. Infatti sovrapponendo diversi orbitali si verifica il fatto che più di due elettroni si verrebbero a trovare con lo stesso set di numeri quantici; l’impossibilità di ciò origina un notevole effetto repulsivo che va a sommarsi a quello originato dalle forze repulsive coulombiane tra i nuclei.
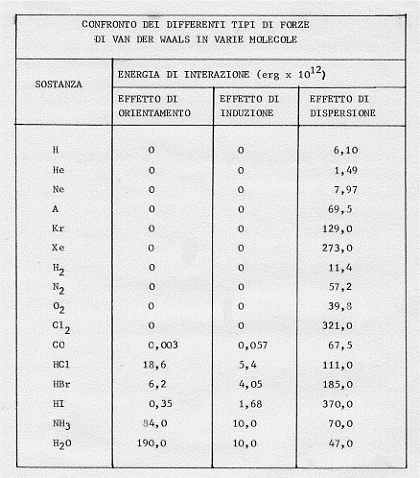
Resta da ultimo da fare il confronto delle forze di orientazione di Keesom, di quelle di induzione di Debye e di quelle di dispersione di London per differenti coppie di atomi e di semplici molecole. Il risultato di questo calcolo fatto da London nel 1937 e da Margenau nel 1939 è riportato nella seguente tavola. Come si può vedere, solo nelle molecole fortemente polari e con piccola polarizzabilità, come H2O, le forze di dispersione sono di secondaria importanza rispetto alle forze di orientamento. Si osservi inoltre che le forze di induzione sono sempre praticamente trascurabili:
In base a quanto abbiamo ora detto e visto, andiamo a disegnare le curve dell’energia potenziale Ep di alcune molecole biatomiche in funzione della distanza r tra i nuclei degli atomi costituenti (vedi figura seguente in cui c’è riportata, per confronto, anche una molecola poliatomica CO2). Il valore del minimo di energia potenziale per le varie molecole indica la diversa stabilità di esse(9):
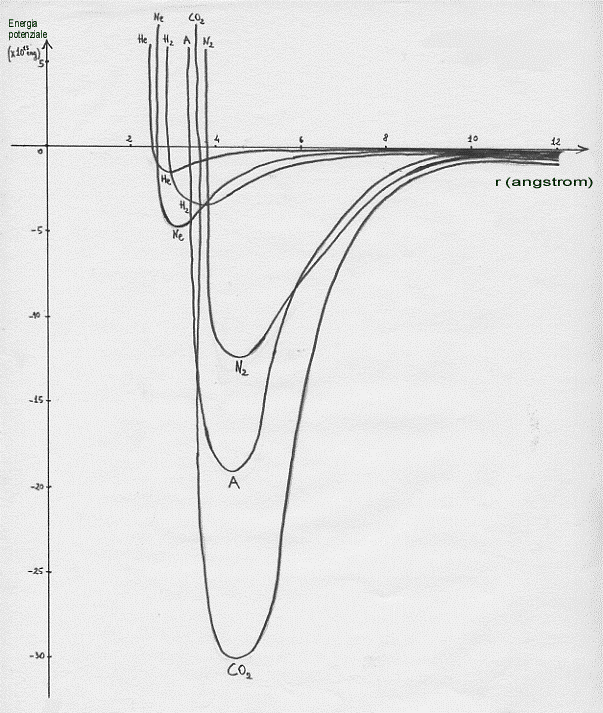
come si può vedere dalla figura la molecola di elio è estremamente instabile; si passa poi a stabilità sempre maggiori fino ad arrivare alla molecola di anidride carbonica (CO2 ) che ha una grande stabilità (relativamente a quella delle altre molecole riportate nel grafico).
***
Prima di chiudere con le forze di Van der Waals occorre trattare un ultimo tipo di interazione, quella che va sotto il nome di legame idrogeno.
NOTE
(1) Lo stesso discorso vale se alle molecole in gioco sono associati dei quadrupoli od ottupoli. Come esempio macroscopico di oscillatore armonico unidimensionale (con un solo grado di libertà) si può considerare una massa m vincolata mediante una molla ad un sostegno e fatta oscillare lungo l’asse x di figura (questa massa è soggetta alla sola forza di richiamo della molla):
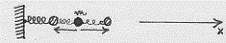
C’è da notare che la quantizzazione dell’oscillatore di figura non è assolutamente rivelabile; solo in scala atomica ha senso polare di quantizzazione.
(2) I successivi valori dell’energia per un oscillatore armonico sono dati dalla formula generale En = (n + ½) hn (con n = 0, 1, 2, …); questi valori saranno quindi in successione crescente: E0 = ½ hn; E1 = 3/2 hn; E2 = 5/2 hn; … Il temine E0 è chiamato energia di punto zero perché corrisponde ad n=0. Supponiamo di avere una massa m, legata ad una molla vincolata ad un sostegno, e di farla oscillare lungo l’asse x di figura:

La forza cui è soggetta la molla, in accordo con la legge di Hooke, è F = -Kx dove K è la costante elastica di richiamo della molla (che dipende dalla natura del materiale di cui essa è costituita) che ha le dimensioni di una massa divisa per un tempo al quadrato. Si può dimostrare che l’energia potenziale che compete alla molla è Ep = ½ Kx2 . Ora, osservando che l’espressione ora fornita rappresenta sugli assi Ep ed x una parabola passante per l’origine, si ha la figura (a) seguente:
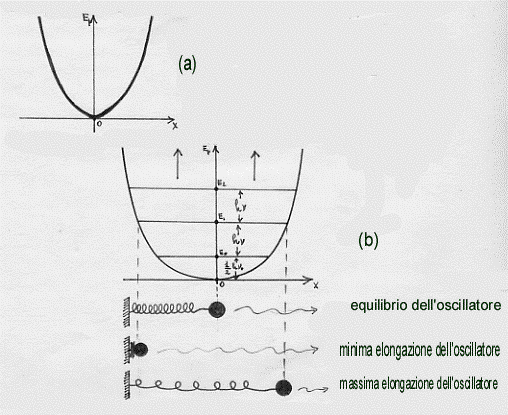
Evidentemente lo stesso discorso vale per le vibrazioni armoniche degli atomi nelle moleco le. Riferiamo allora la figura (a) a queste vibrazioni. Ora, per quanto dicevamo all’inizio della nota, non tutti i valori dell’energia sono permessi agli oscillatori armonici nella molecola, ma solo quelli dati dalla formula En = (n + ½) hn. Vediamo allora quali sono questi valori En dell’energia nella figura (b) precedente. Come si vede dalla figura, il minimo valore dell’energia E di un oscillatore, classicamente, dovrebbe essere zero, mentre quantisticamente è E0. E, mentre classicamente per l’energia Ep sono possibili tutti i valori da 0 ad ∞ , quantisticamente sono possibili solo i valori E0, E1, E2, E3, etc… E’ importante notare e ricordare a parte che, secondo l’ipotesi di Planck, l’energia è ceduta od acquistata per quanti indivisibili hn di energia. Ora la caratteristica peculiare dell’energia di punto zero è che, essendo essa ½ hn, cioè mezzo quanto di Planck, non può essere in alcun modo ceduta dall’oscillatore ma deve essere conservata appunto come lo stato di energia minima.
(3) Fino ad ora abbiamo discusso di un oscillatore unidimensionale, cioè di un oscillatore con un solo grado di libertà, e abbiamo visto il suo valore di energia di punto zero . Nel caso generale di oscillatori tridimensionali (con tre gradi di libertà) l’energia di punto zero è data da 3.(½ hn) = 3/2 hn . Ad ogni grado di libertà compete quindi un’energia di punto zero pari a ½ hn . La formula generale per i livelli energetici permessi all’oscillatore diventa allora:
En = (n + 3/2) hn (con n=0,1,2, ….).
(4) London identificò il termine hn0, che compare nella formula scritta, con il potenziale di ionizzazione dell’atomo o della molecola che si sta considerando come oscillatore.
(5) Una prima intuizione sull’origine di queste forze fu di Wang (1927) il quale calcolò con l’ausilio dell’appena nata meccanica quantistica l’azione che si esercita, a grandi distanze, tra due atomi di idrogeno. Wang trovò che le forze di interazione sono attrattive e variano come 1/r7 (mentre l’energia di interazione varia, come trovato poi da London, come 1/r6) .
(6) La ragione di questa denominazione deriva dal fatto che un metodo di calcolo teorico di queste forze fa intervenire i fenomeni della dispersione della rifrazione.
(7) II linguaggio è semiclassico in quanto il principio di indeterminazione impedisce concettualmente queste possibilità.
(8) Questi dipoli sono naturalmente i dipoli istantanei che abbiamo menzio nato qualche pagina indietro. Va aggiunto qui che questi dipoli cambia no configurazione continuamente e periodicamente nel tempo in accordo con il fatto che gli elettroni degli atomi sono in continuo movimento.
(9) Si ricordi che la distanza tra il minimo di energia e lo zero di questa energia indica l’energia necessaria a dissociare la molecola.
Categorie:Atomi e Molecole

Rispondi